MAF文件格式被广泛用于检测到的体细胞变异。TCGA已经对30多种不同的癌症进行了测序,每种癌症类型的样本量超过200个,由体细胞变异组成的结果数据以突变注释格式的形式保存。maftools试图以一种有效的方式从TCGA来源或其他基因组数据来总结,分析,注释和可视化MAF文件。

1. 安装maftools
#从Bioconductor安装
BiocManager::install("maftools")
#从github安装
BiocManager::install("PoisonAlien/maftools")2. 准备MAF文件
MAF文件生成取决于我们用什么软件进行注释,不同的注释软件生成的VCF文件略有不同。
1)使用VEP注释,可以使用vcf2maf来生成MAF文件
2)使用gatk的Funcotator来注释,可以通过指定参数--output-file-format MAF来生成MAF文件
3)使用ANNOVAR进行注释,可以用过annovarToMaf来生成MAF文件
文件格式介绍如下:
| File formats | Data Portals | Annotation tools |
|---|---|---|
| Mutation Annotation Format(MAF) | TCGA | vcf2maf – for converting your VCF files to MAF |
| Variant Call Format(VCF) | ICGC | Ensembl Variant Effect Predictor VEP |
| ICGC Simple Somatic Mutation Format | Broad Firehose | Annovar |
| cBioPortal | Funcotator | |
| CIViC – Clinical interpretation of variants in cancer | ||
| DGIdb – Information on drug-gene interactions and the druggable genome |
3.maftools输入文件准备
read.maf(
maf,
clinicalData = NULL,
rmFlags = FALSE,
removeDuplicatedVariants = TRUE,
useAll = TRUE,
gisticAllLesionsFile = NULL,
gisticAmpGenesFile = NULL,
gisticDelGenesFile = NULL,
gisticScoresFile = NULL,
cnLevel = "all",
cnTable = NULL,
isTCGA = FALSE,
vc_nonSyn = NULL,
verbose = TRUE
)1)前面提到的MAF文件(可以是gz压缩后的MAF文件, 必须)
2)MAF中与每个Sample/Tumor_Sample_Barcode相关的临床数据(tsv文件格式,可选但推荐,后续可视化可能会用到相关标签)
3)拷贝数数据(如果可用)。可以是GISTIC输出,也可以是包含样本名称、基因名称和拷贝状态(Amp或Del)的数据表。
4.简单展示
此处简单展示maftools的可视化应用,详细内容可以查看官方文档(推荐)
library(maftools)
laml = read.maf(maf = system.file('extdata', 'tcga_laml.maf.gz', package = 'maftools'),
clinicalData = system.file('extdata', 'tcga_laml_annot.tsv', package = 'maftools'))
#查看sample summry.
getSampleSummary(laml)
#查看基因summary.
getGeneSummary(laml)
#查看样本临床信息
getClinicalData(laml)
#查看所有可用的信息
getFields(laml)
#可视化
plotmafSummary(maf = laml, rmOutlier = TRUE, addStat = 'median', dashboard = TRUE, titvRaw = FALSE)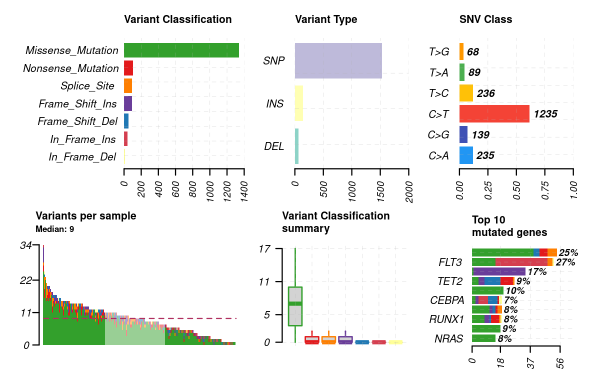
绘制Oncoplots图,这个也是基因组常用的可视化热图
oncoplot(maf = laml, top = 10)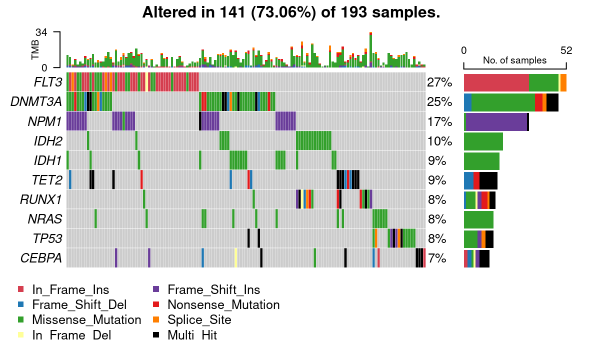
关于maftools的简单介绍就到这里了,下面是一些作者推荐的常用的工具包:
- TRONCO – Repository of the TRanslational ONCOlogy library (R)
- dndscv – dN/dS methods to quantify selection in cancer and somatic evolution (R)
- cloneevol – Inferring and visualizing clonal evolution in multi-sample cancer sequencing (R)
- sigminer – Primarily for signature analysis and visualization in R. Supports
maftoolsoutput (R) - GenVisR – Primarily for visualization (R)
- comut – Primarily for visualization (Python)
- TCGAmutations – pre-compiled curated somatic mutations from TCGA cohorts (from Broad Firehose and TCGA MC3 Project) that can be loaded into
maftools(R) - somaticfreq – rapid genotyping of known somatic hotspot variants from the tumor BAM files. Generates a browsable/sharable HTML report. (C)
参考资料:
1.https://bioconductor.org/packages/release/bioc/vignettes/maftools/inst/doc/maftools.html
2.https://github.com/PoisonAlien/maftools
3.Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. 2018. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Resarch. PMID: 30341162
