
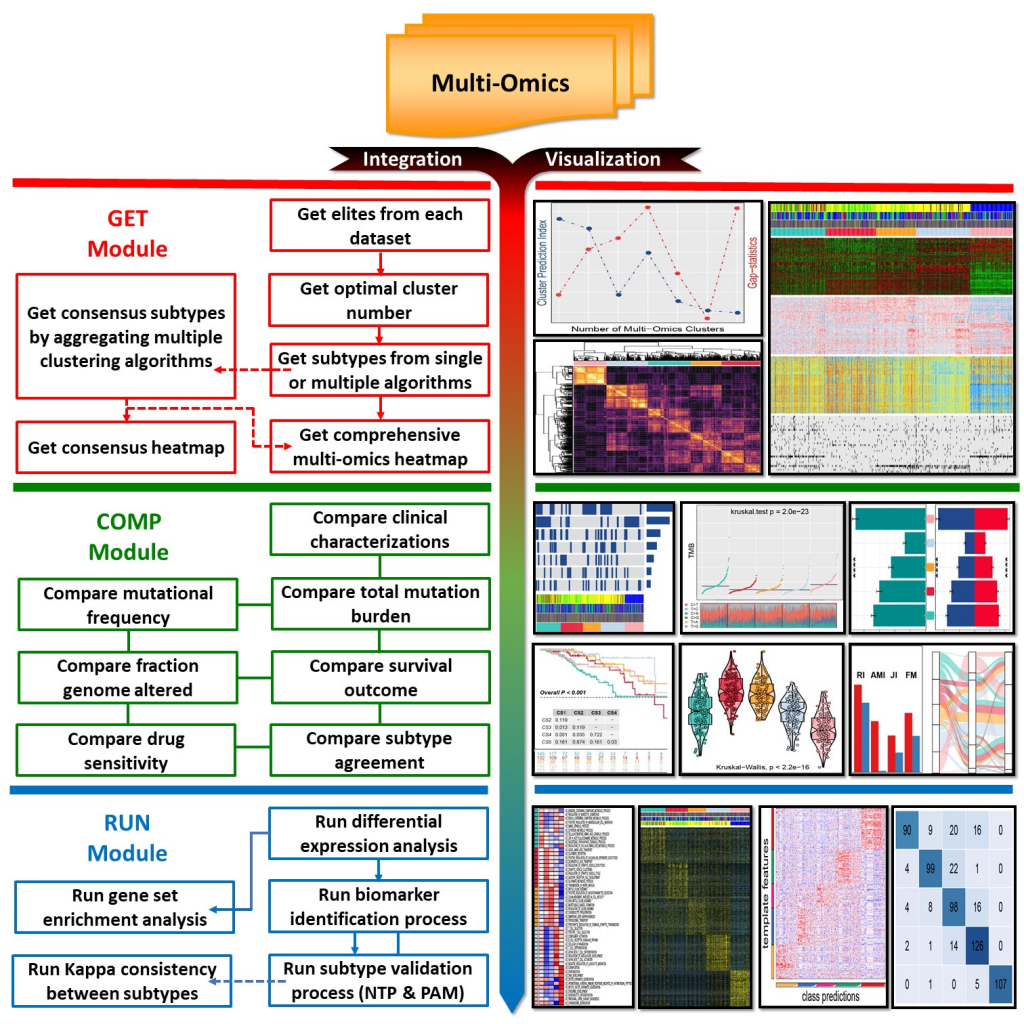
MOVICS是一款整合了多组学数据分析及可视化的R包,整体流程涵盖了多组学分析的诸多方面数据分析包含三大模块:
- GET Module:通过聚类获取多组学数据中的亚型、分类
- COMP Module:多个维度来比较分型结果
- RUN Module:分型结果验证和Mark挖掘
具体见下图:
1)R包安装
首先确保你的R版本 > 4.0.1且BiocManager v3.11然后在进行安装,版本不对应可能会导致安装失败,同时依赖的安装包也比较多:CIMLR,ClassDiscovery,ConsensusClusterPlus,IntNMF,PINSPlus,SNFtool,coca,dplyr,ggplot2,iClusterPlus,mogsa,vegan,circlize,survival,survminer,ggpp,tibble,limma,DESeq2,edgeR,aricode,ggalluvial,flexclust,reshape2,clusterProfiler,GSVA,grid,cowplot,jstable,impute,CMScaller,car,genefilter,ggpubr,preprocessCore,ridge,sva,grDevices,maftools,patchwork,ComplexHeatmap (>= 2.5.5),pamr,clusterRepro,officer。请确认依赖包都安装好后安装:
# 利用devtools安装GitHub中托管的R包
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
if (!require("devtools"))
install.packages("devtools")
# 安装包比较大,下载需要时间
devtools::install_github("xlucpu/MOVICS")几个下载困难的包已经上传云盘,方便下载:云盘下载
下载后可以直接按照下面命令运行安装:
install.packages("D:/aricode_1.0.1.tar.gz", repos = NULL, type = "source")2)示例演示
我们利用官方的文档数据进行演示说明
library(MOVICS)
# 作者提供的演示数据
load(system.file("extdata", "brca.tcga.RData", package = "MOVICS", mustWork = TRUE))
load(system.file("extdata", "brca.yau.RData", package = "MOVICS", mustWork = TRUE))
# 提取数据用于下游分析
mo.data <- brca.tcga[1:4]
count <- brca.tcga$count
fpkm <- brca.tcga$fpkm
maf <- brca.tcga$maf
segment <- brca.tcga$segment
surv.info <- brca.tcga$clin.info获取最优分类数,getClustNum函数利用CPI5和Gaps-statistics方法来获取最优的聚类数:
optk.brca <- getClustNum(data = mo.data,
is.binary = c(F,F,F,T), # 第四组数据是somatic mutation数据,属于binary matrix
try.N.clust = 2:8, # 从2个亚类尝试直到8个亚类,可以自定义
fig.name = "CLUSTER NUMBER OF TCGA-BRCA")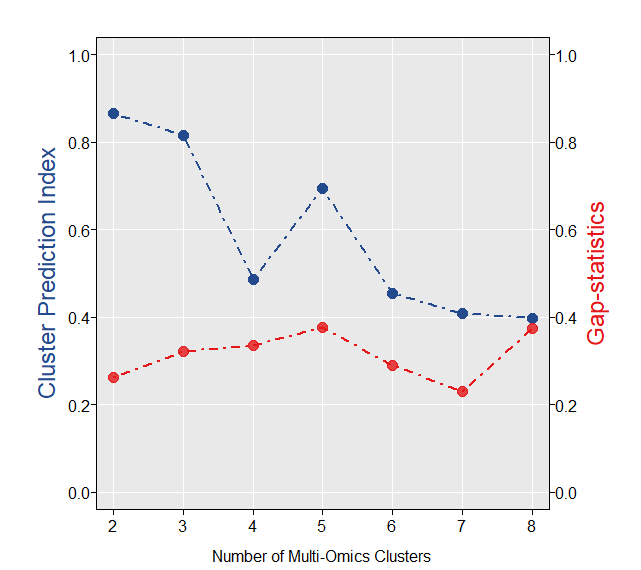
从图上简单来看,当聚类数为5时为最优(也可以结合具体case的生物学意义进行选择)
MOVICS提供了多种聚类算法供我们选择,调用方法也比较灵活,如下:
# 假设我们需要使用iClusterBayes 聚类算法我们可以使用如下方式调用:
iClusterBayes.res <- getiClusterBayes(data = mo.data,
N.clust = 5,
type = c("gaussian","gaussian","gaussian","binomial"),
n.burnin = 1800,
n.draw = 1200,
prior.gamma = c(0.5, 0.5, 0.5, 0.5),
sdev = 0.05,
thin = 3)
# 也可以用使用getMOIC函数指定methodslist聚类方法,如下:
iClusterBayes.res <- getMOIC(data = mo.data,
N.clust = 5,
methodslist = "iClusterBayes", # specify only ONE algorithm here
type = c("gaussian","gaussian","gaussian","binomial"), # data type corresponding to the list
n.burnin = 1800,
n.draw = 1200,
prior.gamma = c(0.5, 0.5, 0.5, 0.5),
sdev = 0.05,
thin = 3)同时我们也可以一次调用多种聚类算法,不过这样需要花费很长时间:
moic.res.list <- getMOIC(data = mo.data,
methodslist = list("SNF", "PINSPlus", "NEMO", "COCA", "LRAcluster", "ConsensusClustering", "IntNMF", "CIMLR", "MoCluster"),
N.clust = 5,
type = c("gaussian", "gaussian", "gaussian", "binomial"))最后,使用getConsensusMOIC获取聚类结果热图
cmoic.brca <- getConsensusMOIC(moic.res.list = moic.res.list,
fig.name = "CONSENSUS HEATMAP",
distance = "euclidean",
linkage = "average")
然后我们利用定量结果的相似性评价分类的效果
getSilhouette(sil = cmoic.brca$sil, # a sil object returned by getConsensusMOIC()
fig.path = getwd(),
fig.name = "SILHOUETTE",
height = 5.5,
width = 5)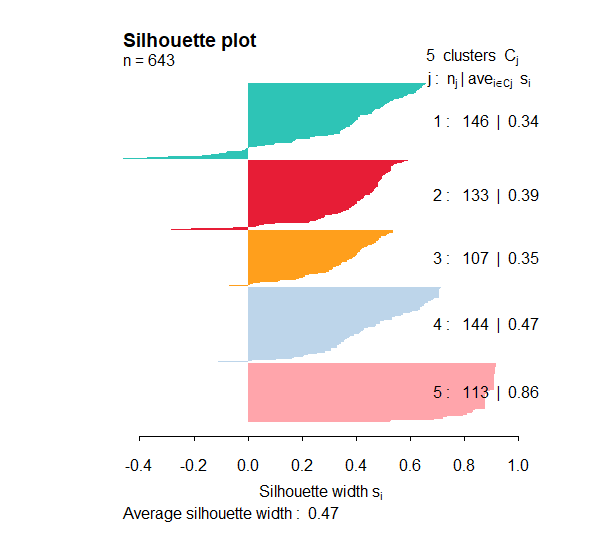
最后我们根据聚类结果绘制热图
# 将甲基化数据的 beta value转化为 M value
indata <- mo.data
indata$meth.beta <- log2(indata$meth.beta / (1 - indata$meth.beta))
# 数据标准化
plotdata <- getStdiz(data = indata,
halfwidth = c(2,2,2,NA), # no truncation for mutation
centerFlag = c(T,T,T,F), # no center for mutation
scaleFlag = c(T,T,T,F)) # no scale for mutation
# 选取前10个feature
feat <- iClusterBayes.res$feat.res
feat1 <- feat[which(feat$dataset == "mRNA.expr"),][1:10,"feature"]
feat2 <- feat[which(feat$dataset == "lncRNA.expr"),][1:10,"feature"]
feat3 <- feat[which(feat$dataset == "meth.beta"),][1:10,"feature"]
feat4 <- feat[which(feat$dataset == "mut.status"),][1:10,"feature"]
annRow <- list(feat1, feat2, feat3, feat4)
# 定义不同组学颜色展示
mRNA.col <- c("#00FF00", "#008000", "#000000", "#800000", "#FF0000")
lncRNA.col <- c("#6699CC", "white" , "#FF3C38")
meth.col <- c("#0074FE", "#96EBF9", "#FEE900", "#F00003")
mut.col <- c("grey90" , "black")
col.list <- list(mRNA.col, lncRNA.col, meth.col, mut.col)
# 绘图,较慢
getMoHeatmap(data = plotdata,
row.title = c("mRNA","lncRNA","Methylation","Mutation"),
is.binary = c(F,F,F,T), # the 4th data is mutation which is binary
legend.name = c("mRNA.FPKM","lncRNA.FPKM","M value","Mutated"),
clust.res = iClusterBayes.res$clust.res, # cluster results
clust.dend = NULL, # no dendrogram
show.rownames = c(F,F,F,F), # specify for each omics data
show.colnames = FALSE, # show no sample names
annRow = annRow, # mark selected features
color = col.list,
annCol = NULL, # no annotation for samples
annColors = NULL, # no annotation color
width = 10, # width of each subheatmap
height = 5, # height of each subheatmap
fig.name = "COMPREHENSIVE HEATMAP OF ICLUSTERBAYES")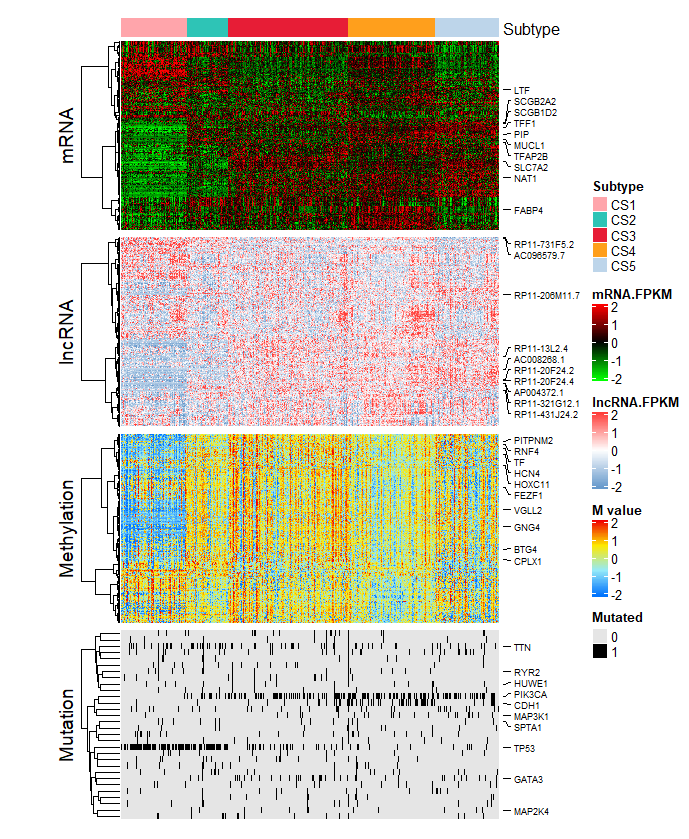
关于MOVICS的介绍就到这里了,该包还提供了很多可视化方法,详细见参考资料。
参考资料:
1.https://github.com/xlucpu/MOVICS
2.https://xlucpu.github.io/MOVICS/MOVICS-VIGNETTE.html
3.Lu, X., Meng, J., Zhou, Y., Jiang, L., and Yan, F. (2020). MOVICS: an R package for multi-omics integration and visualization in cancer subtyping. Bioinformatics, btaa1018.
