
MCScanX是检测基因共线性和进化分析的常用的分析工具之一,利用两个物种蛋白质blastp比对结果,再结合这些蛋白质基因在基因组中的位置,得到两个物种基因组的共线性区块。

安装编译:
# 下载:http://chibba.pgml.uga.edu/mcscan2/MCScanX.zip
unzip MCscanX.zip && cd MCScanX && make注意:如果编译报错:getopt未定义,需要添加#include <unistd.h>到相应报错的.h文件中。
教程示例:
| 程序名称 | 物种 | 计算花费时间 |
| MCScanX | Arabidopsis (at) | <1 |
| MCScanX | Arabidopsis (at) and grape (vv) | <1 |
| MCScanX_h | Arabidopsis (at), soybean (gm), poplar (pt) and grape (vv) | ~2 |
| duplicate_gene_classifier | Arabidopsis (at) | <1 |
| detect_collinear_tandem_arrays | Arabidopsis (at) and grape (vv) | <1 |
| dissect_multiple_alignments | Arabidopsis (at) and grape (vv) | <1 |
| dot_plotter, dual_synteny_plotter, circle_plotter and bar_plotter | Rice (os) and sorghum (sb) | <1 |
| add_ka_and_ks_to_collinearity | Arabidopsis (at) | ~5 |
| group_collinear_genes | Arabidopsis (at) | <1 |
| detect_collinearity_within_gene_families | Arabidopsis (at) | <1 |
| origin_enrichment_analysis | Arabidopsis (at) | <1 |
| family_circle_plotter | Arabidopsis (at) | <1 |
| family_tree_plotter | Arabidopsis (at) | <1 |
输入文件主要是blastp结果文件和相应的gff注释文件(注意文件格式,可以根据官方示例学习使用方法)。
作者也提供了很多下游分析的绘图脚本( 需要 java环境):
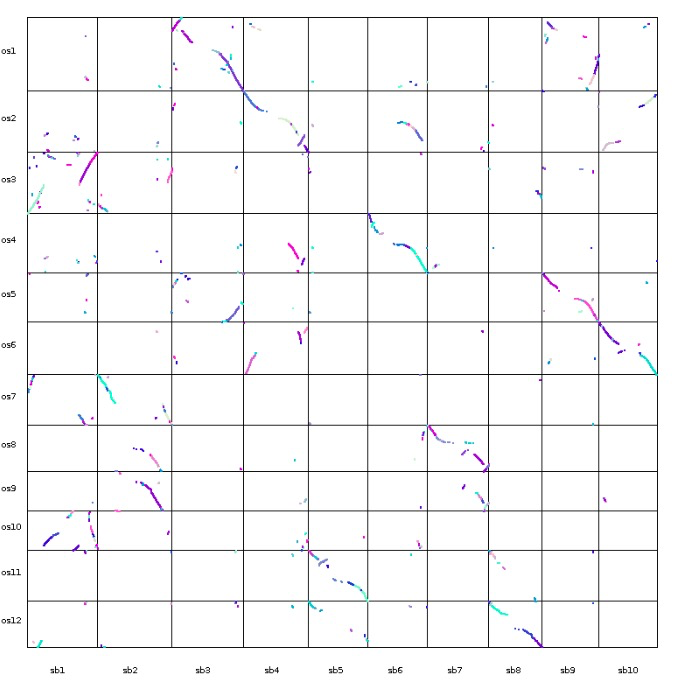
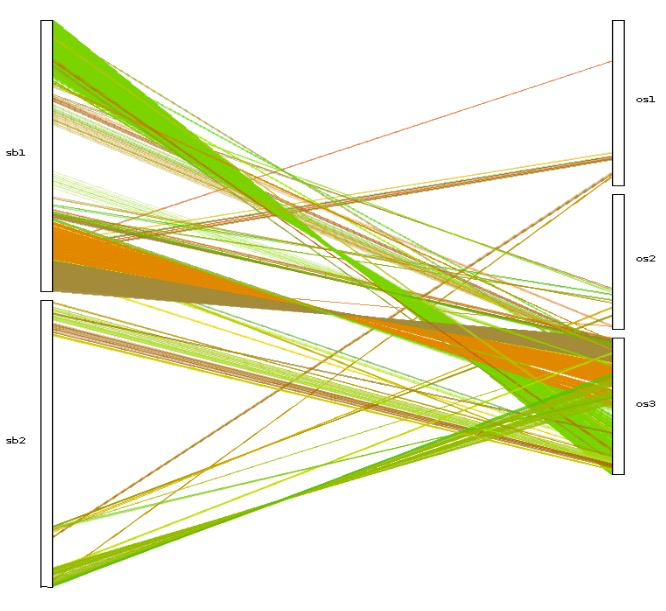
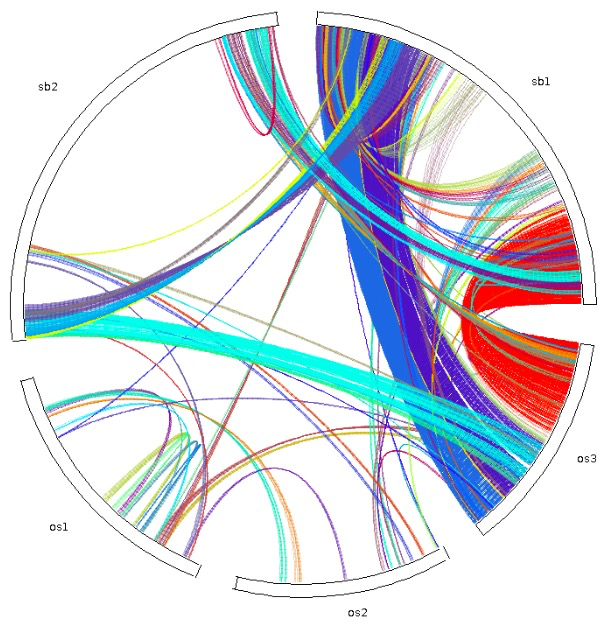
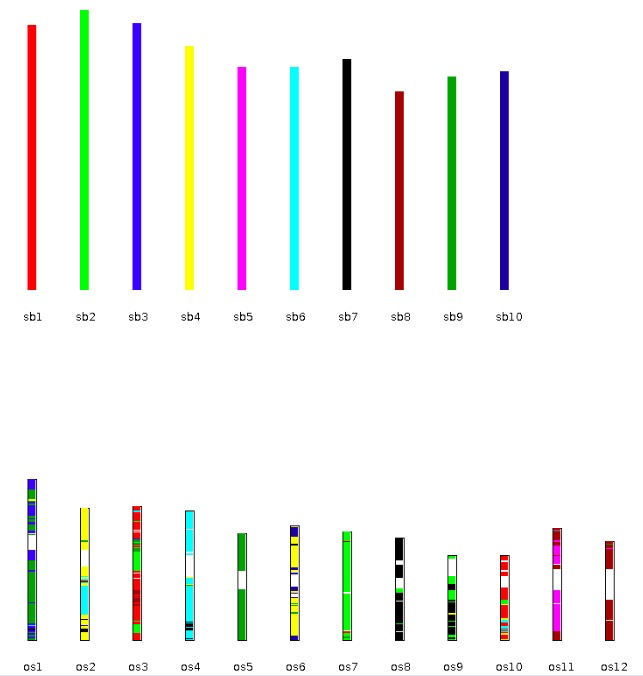
更多内容可以参考官方文档,基本都是一行命令,对生信人员来说使用起来非常简单。
参考资料:
1.http://chibba.pgml.uga.edu/mcscan2/index.php#ex
