
一般我们常用R中Seurat、scran、monocle等的R包来处理单细胞数据,但是R在读取和处理数据的过程中会将所有的变量和占用都储存在RAM当中,在处理海量数据时经常会出现内存不足的情况。
scanpy是一个基于python的单细胞基因表达分析包,它包含了数据预处理、可视化、聚类、伪时间分析和轨迹推断、差异表达分析等数据分析手段等。
The Python-based implementation efficiently deals with datasets of more than one million cells.
https://scanpy.readthedocs.io/en/stable/index.html
安装scanpy:
# 建议用conda安装, 建议python版本大于3.6
# 安装依赖包
conda install seaborn scikit-learn statsmodels numba pytables
conda install -c conda-forge python-igraph leidenalg
# 安装scanpy,目前好像这个用conda还无法完成正确安装
pip install scanpyDocker:docker pull fastgenomics/scanpy
教程:
1)聚类:https://scanpy-tutorials.readthedocs.io/en/latest/pbmc3k.html
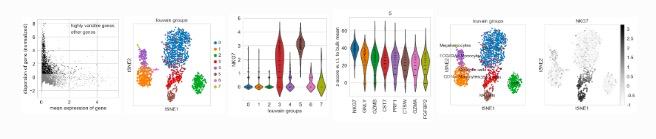
2)可视化:https://scanpy-tutorials.readthedocs.io/en/latest/plotting/core.html

3)轨迹推断:https://scanpy-tutorials.readthedocs.io/en/latest/paga-paul15.html


4)数据整合分析(整合、降维、注释等):https://scanpy-tutorials.readthedocs.io/en/latest/integrating-data-using-ingest.html

5)空间分数分析:https://scanpy-tutorials.readthedocs.io/en/latest/spatial/basic-analysis.html
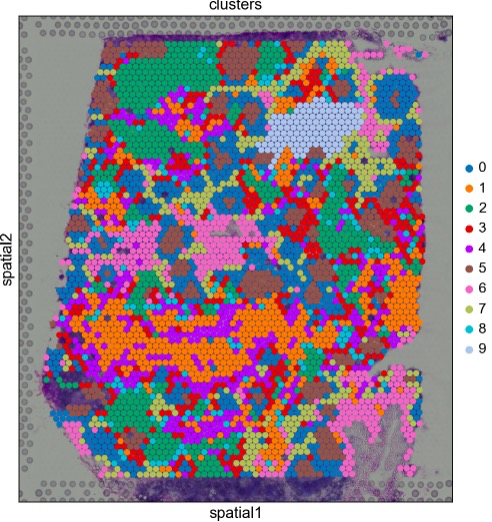
关于scanpy的介绍就到这里了,后面可以简单演示一下其分析流程和思路。
参考资料:
1.https://scanpy.readthedocs.io/en/stable/installation.html
