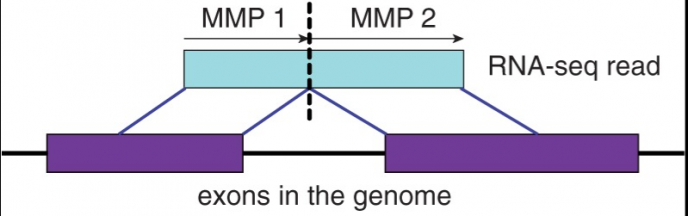
前面一篇提到《RNA-seq数据分析全流程(思路篇)》,本文主要依据上面的思路做一个代码流程的分享,主要参考GDC的数据分析流程:
1)序列比对
# STAR-2.6.0c
# 安装STAR:https://github.com/alexdobin/STAR
# 文档:https://github.com/alexdobin/STAR/blob/master/doc/STARmanual.pdf
STAR \
--readFilesIn <fastq_files> \
--outSAMattrRGline <read_group_strings> \
--alignIntronMax 1000000 \
--alignIntronMin 20 \
--alignMatesGapMax 1000000 \
--alignSJDBoverhangMin 1 \
--alignSJoverhangMin 8 \
--alignSoftClipAtReferenceEnds Yes \
--chimJunctionOverhangMin 15 \
--chimMainSegmentMultNmax 1 \
--chimOutType Junctions SeparateSAMold WithinBAM SoftClip \
--chimSegmentMin 15 \
--genomeDir <genome_dir> \
--genomeLoad NoSharedMemory \
--limitSjdbInsertNsj 1200000 \
--outFileNamePrefix <output_prefix> \
--outFilterIntronMotifs None \
--outFilterMatchNminOverLread 0.33 \
--outFilterMismatchNmax 999 \
--outFilterMismatchNoverLmax 0.1 \
--outFilterMultimapNmax 20 \
--outFilterScoreMinOverLread 0.33 \
--outFilterType BySJout \
--outSAMattributes NH HI AS nM NM ch \
--outSAMstrandField intronMotif \
--outSAMtype BAM Unsorted \
--outSAMunmapped Within \
--quantMode TranscriptomeSAM GeneCounts \
--readFilesCommand <zcat, etc> \
--runThreadN <threads> \
--twopassMode Basic2)计算mRNA count值(此处我们用htseq,也可以用)
# 安装:https://htseq.readthedocs.io/en/master/install.html
# 介绍文档:https://htseq.readthedocs.io/en/master/count.html#count
htseq-count \
-f bam \
-r name \
-s no \
-a 10 \
-t exon \
-i gene_id \
-m intersection-nonempty \
<input_bam> \
<gtf_file> > <counts_file>
3)mRNA-Seq表达值标准化
一般我们比较常用的是FPKM值进行计算。

4)差异分析
可以参考我之前的文章《三种不同的T-Test介绍》根据不同的情况来进行求差异蛋白。
在软件选用上推荐用limma、DESeq2、edgeR等来做分析,一般设置的阈值为|log2FC| > 1和FDR < 0.05的条件进行过滤差异基因,如果针对数据质量或者测序深度较少的情况也可以用 |log2FC| > 1和P < 0.05 的条件进行差异分析,具体情况具体对待。
5)下游分析
此处涉及功能、通路分析、关联分析等内容,大家可以搜素本博客之前分享的内容,未介绍的内容未来大家分享。
参考资料:
1.https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
2.https://pubmed.ncbi.nlm.nih.gov/23104886/
