Spectronaut软件作为蛋白组DIA分析商用最常用的谱图分析软件,其主要特点:
- 深度整合深度学习方法,最新的14版本采用新的
directDIA 2.0library free引擎,使其鉴定率大大提高 - 支持最新的
Thermo FAIMS Pro、Bruker dia-PASEF等最新的质谱方法 - 对队列数据分析做了诸多的改进,比如支持分批次分析然后合并(当然在服务器够用的情况还是推荐一次性跑完)
- 基于windows(目前没有linux版),对用户操作友好,但是对于我们常做队列数据分析来说不太友好

优势方面就不多介绍了,在蛋白质组学它几乎成了DIA数据分析的金标准,当然价格不菲( 可能这是其主要缺点吧)。官方支持试用,需要发邮件申请,他们回复基本很快。
1)首先,我们安装好 Spectronaut后需要配置缓存目录,这个非常重要,软件默认是C盘,这个很快会占满,建议分配到较大的盘符(最好是固态硬盘)
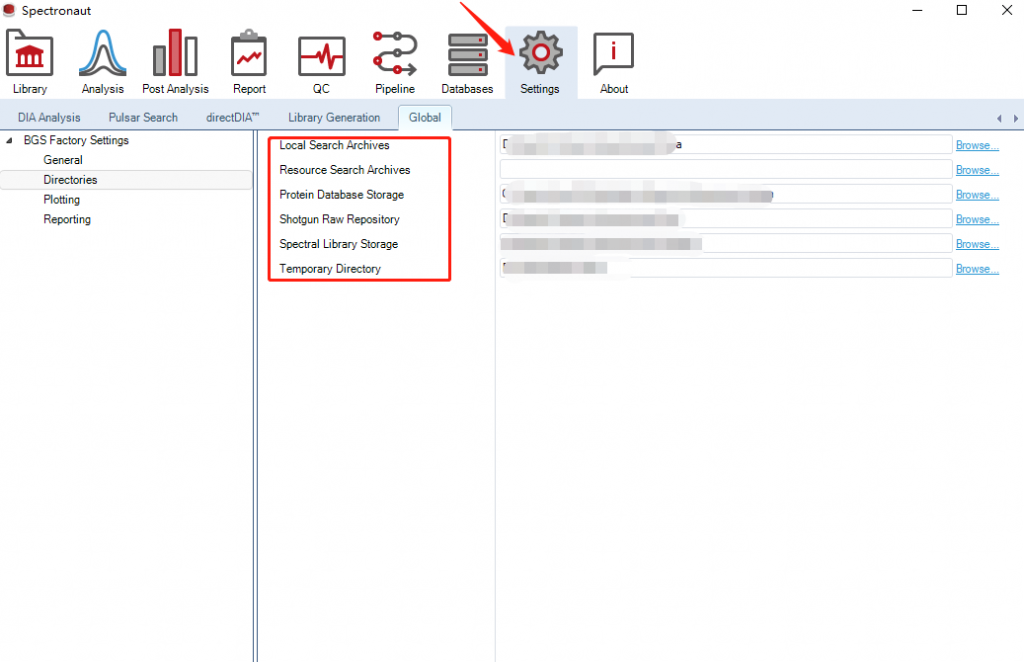
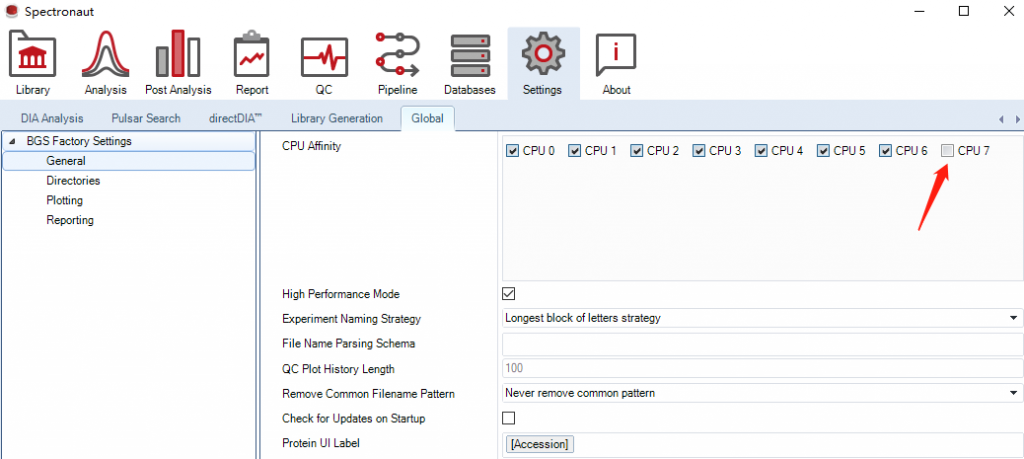
另一个比较重要的设置是CPU使用,默认是占满的。但是有时我们还同时处理其他的程序或者操作,为了避免卡死建议留一个线程。
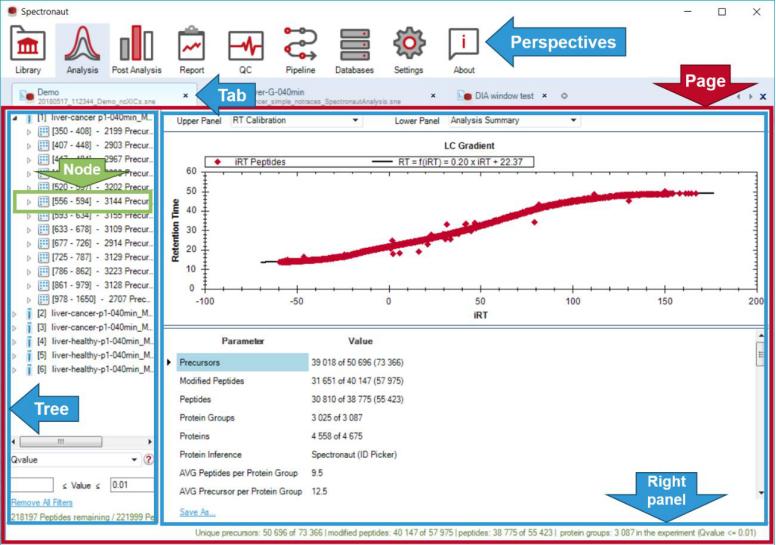
2)程序布局方面也很简洁
3)以一个基于library的DIA数据分析为例
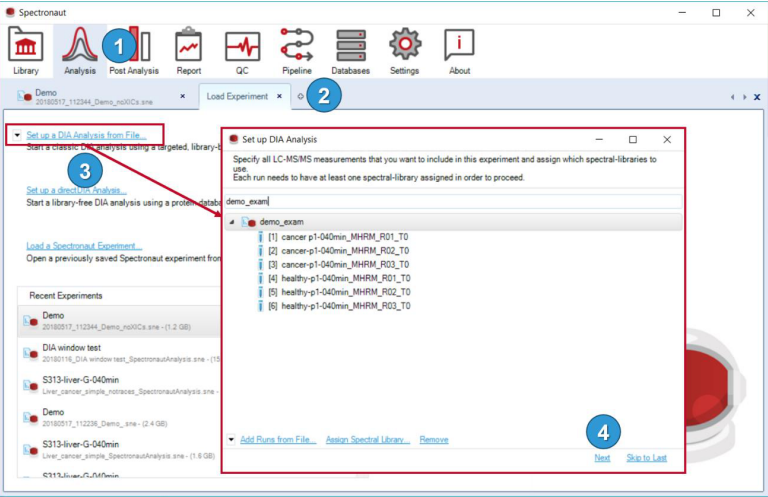
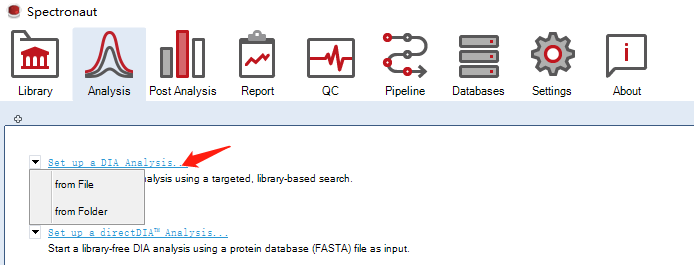
首先点击Assign Spectral Library…可以选择文件夹或者单个文件
然后点击 next 添加Library,也可以添加已经建好的DDA库
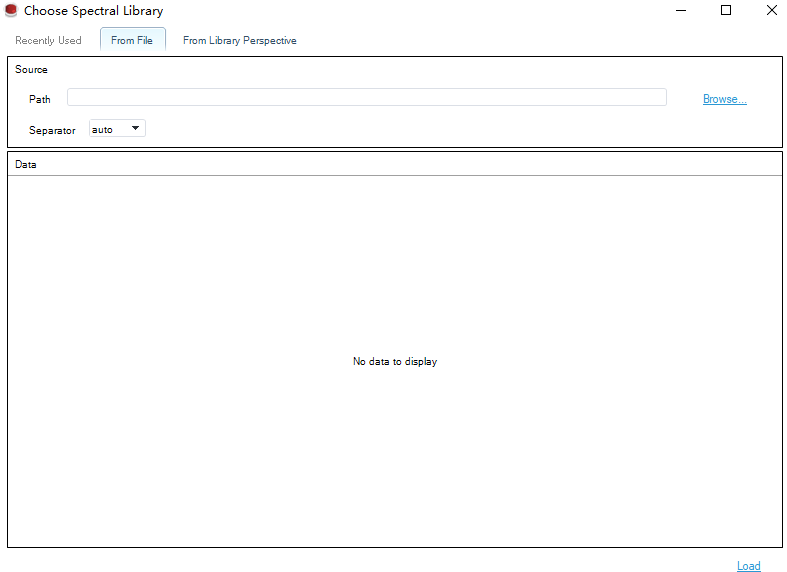
点击Load后进入分析设置页面
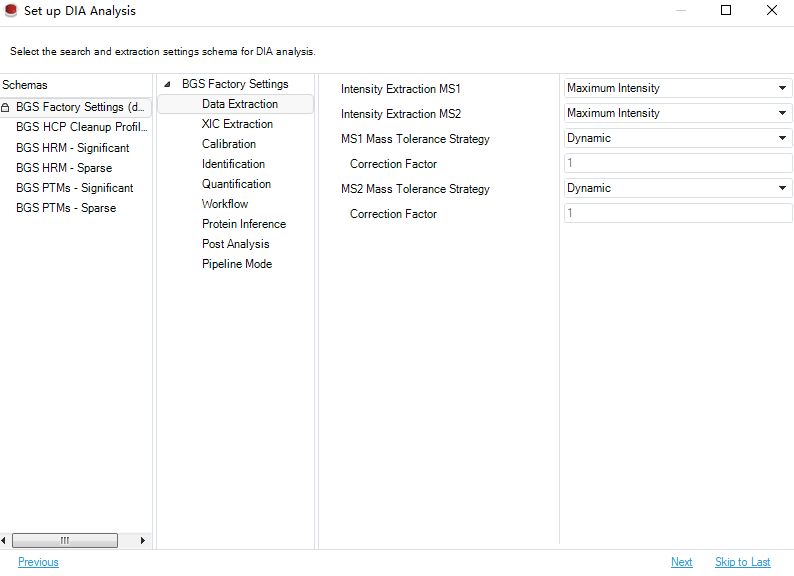
基本上软件会自动判断,一般默认是Dynamic,不用修改。点击Next,会进入fasta文件选择界面,选择合适的 fasta 即可。
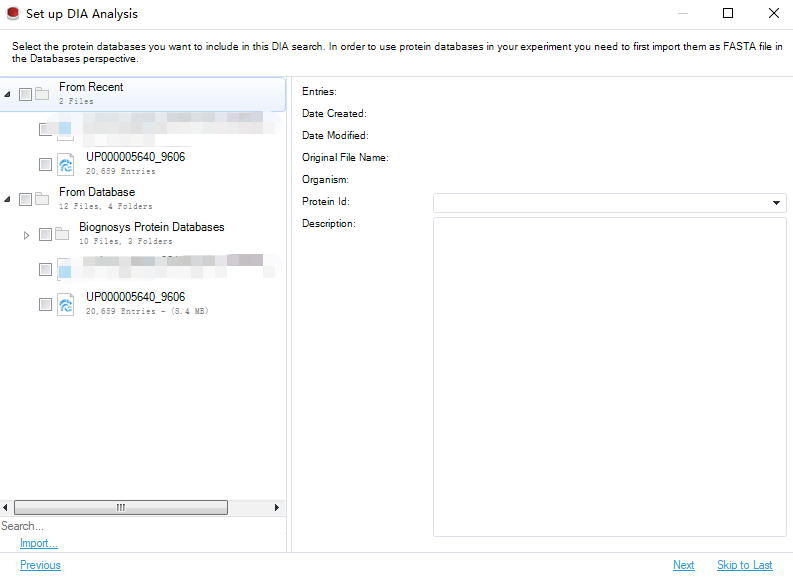
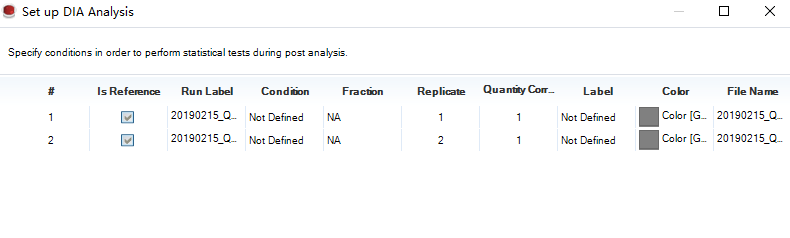
下一步,即设计实验,根据你的分组进行修改:
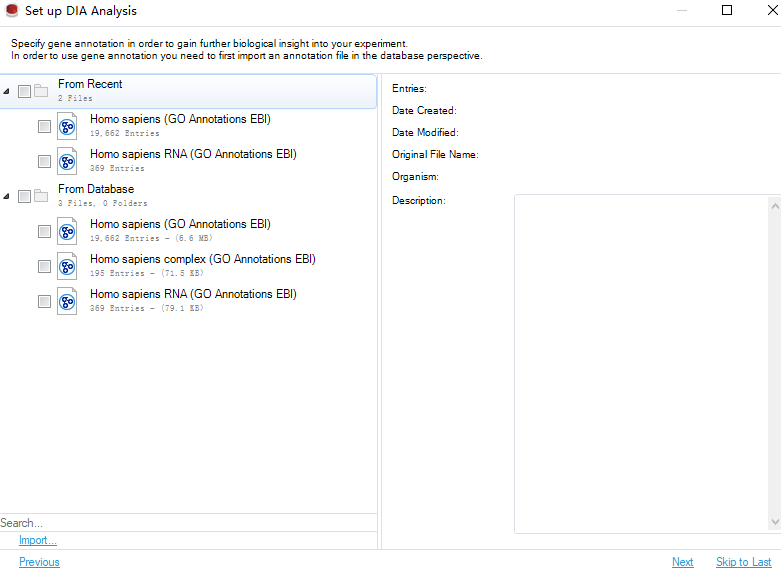
设置完成后进行下一步,选择GO数据库:
最后点击下一步,最后点击Finish即可。
最后有一点需要注意,数据分析完成后记得保存为sne未来可以重复利用和筛选,否则需要重新分析了!!!
参考资料:
1.https://biognosys.com/shop/spectronaut
