
最近研究从基因组数据生成质朴数据搜库个性化fasta文件,产生这样的思考主要是:
1)想利用同一个个体的组学数据研究疾病的发生发展,到最后的蛋白的动态变化,如果我们利用标准的uniport或者swissport数据会遗漏一些特有的碱基变异等信息,我们是否可以利用同一个人的组学数据生成一个fasta?
2)如果做蛋白质组学物种的数据公共数据库中没有其参考的fasta文件如何做?是否可以利用基因组de novo的方法或者说直接利用蛋白质组de novo拼接?
基于以上问题查阅了相关的资料,发现在13年左右已经有人提出了相关的解决方案,利用转录组数据生成fasta并制作了一个小工具在R biocondutor上,在学习之余也和大家分享,以下是其主要分析流程:
# 安装
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
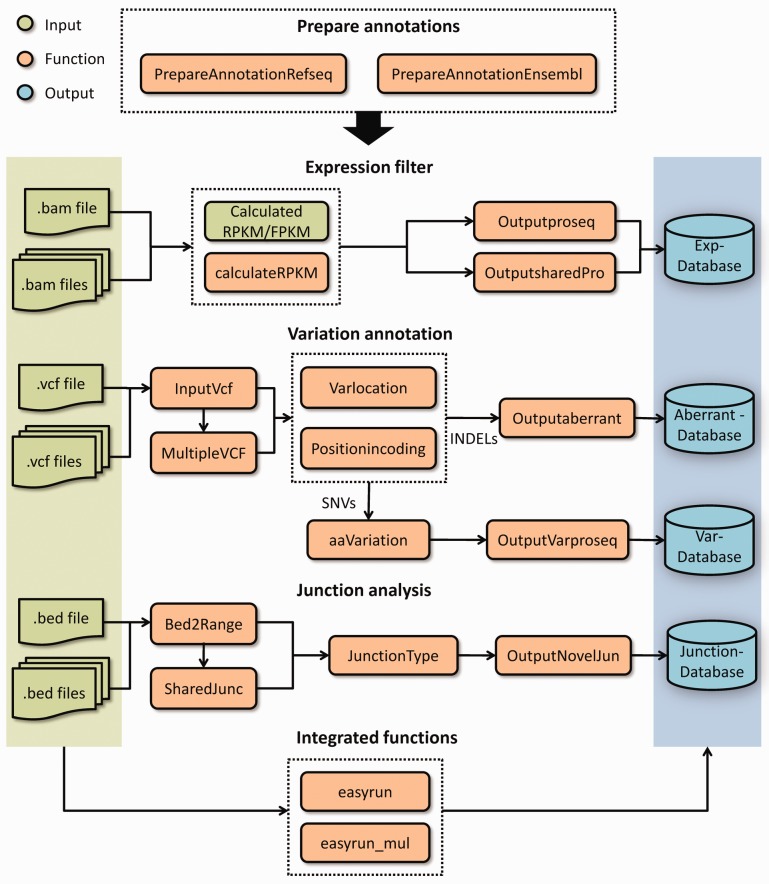
BiocManager::install("customProDB")通过文章我们可以看到主要包含四块内容:
1)基于FPKM/RPKM过滤后基于表达数据生成的fasta文件
示例
load(system.file("extdata/refseq", "exon_anno.RData", package="customProDB"))
load(system.file("extdata/refseq", "proseq.RData", package="customProDB"))
bamFile <- system.file("extdata/bams", "test1_sort.bam",
package="customProDB")
load(system.file("extdata/refseq", "ids.RData", package="customProDB"))
RPKM <- calculateRPKM(bamFile, exon, proteincodingonly=TRUE, ids)
outf1 <- paste(tempdir(), '/test_rpkm.fasta', sep='')
Outputproseq(RPKM, 1, proteinseq, outf1, ids)输出多个样本中高表达的序列
path <- system.file("extdata/bams", package="customProDB")
load(system.file("extdata/refseq", "exon_anno.RData", package="customProDB"))
load(system.file("extdata/refseq", "proseq.RData", package="customProDB"))
load(system.file("extdata/refseq", "ids.RData", package="customProDB"))
bamFile<- paste(path, '/', list.files(path, pattern="*bam$"), sep='')
rpkms <- sapply(bamFile,function(x)
calculateRPKM(x, exon, proteincodingonly=TRUE, ids))
outfile <- paste(tempdir(), '/test_rpkm_share.fasta', sep='')
OutputsharedPro(rpkms, cutoff=1, share_sample=2, proteinseq,
outfile, ids)2)基于vcf,插入、缺失等一些关联注释生成的fasta文件
示例
vcffile <- system.file("extdata/vcfs", "test1.vcf", package="customProDB")
vcf <- InputVcf(vcffile)
table(values(vcf[[1]])[['INDEL']])
index <- which(values(vcf[[1]])[['INDEL']] == TRUE)
indelvcf <- vcf[[1]][index]
load(system.file("extdata/refseq", "exon_anno.RData", package="customProDB"))
load(system.file("extdata/refseq", "dbsnpinCoding.RData",
package="customProDB"))
load(system.file("extdata/refseq", "procodingseq.RData",
package="customProDB"))
load(system.file("extdata/refseq", "proseq.RData", package="customProDB"))
load(system.file("extdata/refseq", "ids.RData", package="customProDB"))
postable_indel <- Positionincoding(indelvcf, exon)
txlist_indel <- unique(postable_indel[, 'txid'])
codingseq_indel <- procodingseq[procodingseq[, 'tx_id'] %in% txlist_indel, ]
outfile <- paste(tempdir(), '/test_indel.fasta', sep='')
Outputaberrant(postable_indel, coding=codingseq_indel,
proteinseq=proteinseq, outfile=outfile, ids=ids)3)基于vcf,SNV(单核苷酸多态性)生成的fasta文件
示例,输出编码蛋白的序列信息:
vcffile <- system.file("extdata/vcfs", "test1.vcf", package="customProDB")
vcf <- InputVcf(vcffile)
table(values(vcf[[1]])[['INDEL']])
index <- which(values(vcf[[1]])[['INDEL']] == FALSE)
SNVvcf <- vcf[[1]][index]
load(system.file("extdata/refseq", "exon_anno.RData",
package="customProDB"))
load(system.file("extdata/refseq", "dbsnpinCoding.RData",
package="customProDB"))
load(system.file("extdata/refseq", "procodingseq.RData",
package="customProDB"))
load(system.file("extdata/refseq", "ids.RData", package="customProDB"))
load(system.file("extdata/refseq", "proseq.RData", package="customProDB"))
postable_snv <- Positionincoding(SNVvcf, exon, dbsnpinCoding)
txlist <- unique(postable_snv[, 'txid'])
codingseq <- procodingseq[procodingseq[, 'tx_id'] %in% txlist, ]
mtab <- aaVariation (postable_snv, codingseq)
OutputVarprocodingseq(mtab, codingseq, ids, lablersid=TRUE)输出编码蛋白到fasta文件
vcffile <- system.file("extdata/vcfs", "test1.vcf", package="customProDB")
vcf <- InputVcf(vcffile)
table(values(vcf[[1]])[['INDEL']])
index <- which(values(vcf[[1]])[['INDEL']] == FALSE)
SNVvcf <- vcf[[1]][index]
load(system.file("extdata/refseq", "exon_anno.RData",
package="customProDB"))
load(system.file("extdata/refseq", "dbsnpinCoding.RData",
package="customProDB"))
load(system.file("extdata/refseq", "procodingseq.RData",
package="customProDB"))
load(system.file("extdata/refseq", "ids.RData", package="customProDB"))
load(system.file("extdata/refseq", "proseq.RData", package="customProDB"))
postable_snv <- Positionincoding(SNVvcf, exon, dbsnpinCoding)
txlist <- unique(postable_snv[, 'txid'])
codingseq <- procodingseq[procodingseq[, 'tx_id'] %in% txlist, ]
mtab <- aaVariation (postable_snv, codingseq)
outfile <- paste(tempdir(), '/test_snv.fasta',sep='')
snvproseq <- OutputVarproseq(mtab, proteinseq, outfile, ids, lablersid=TRUE, RPKM=NULL)4) 基于bed文件产生的新的转录本、复合体或者剪接体的fasta文件
示例:
bedfile <- system.file("extdata/beds", "junctions1.bed", package="customProDB")
jun <- Bed2Range(bedfile,skip=1,covfilter=5)
load(system.file("extdata/refseq", "splicemax.RData", package="customProDB"))
load(system.file("extdata/refseq", "ids.RData", package="customProDB"))
txdb <- loadDb(system.file("extdata/refseq", "txdb.sqlite",
package="customProDB"))
junction_type <- JunctionType(jun, splicemax, txdb, ids)
table(junction_type[, 'jun_type'])
chrom <- paste('chr',c(1:22,'X','Y','M'),sep='')
junction_type <- subset(junction_type, seqnames %in% chrom)
outf_junc <- paste(tempdir(), '/test_junc.fasta', sep='')
load(system.file("extdata/refseq", "proseq.RData", package="customProDB"))
library('BSgenome.Hsapiens.UCSC.hg19')
OutputNovelJun <- OutputNovelJun(junction_type, Hsapiens, outf_junc,
proteinseq)参考文献:
1. Wang X, Zhang B. customProDB: an R package to generate customized protein databases from RNA-Seq data for proteomics search. Bioinformatics. 2013;29(24):3235-3237. doi:10.1093/bioinformatics/btt543
