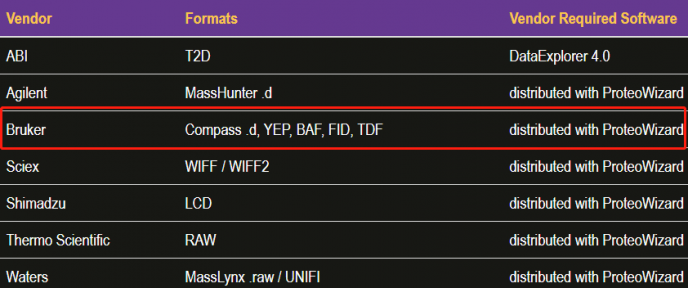
msconvert是一个命令行工具,用于在各种质谱原始文件格式到开放格式(如:mzML、mzXML等)之间进行转换。对于Windows用户,msConvertGUI程序也可用于简单的文件转换。下面我们主要介绍如何将timsTOF raw文件转换成mzML。
下载:链接
然后安装,完成后打开界面如下:

然后按照下面方式添加:

1.选择.d文件夹
2.添加文件夹,多个文件请依次添加
3.指定输出目录
4.选择“Combine ion mobility scans”项
5.添加“scanSumming”和“threshold”,具体修改如下
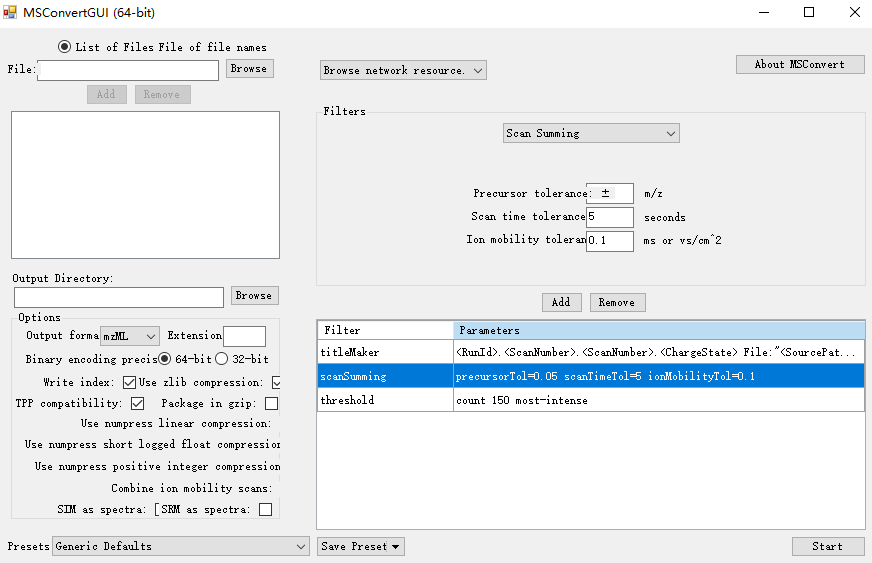

然后点击start开始即可。
命令行版参考:https://evvail.com/2019/12/19/293.html
参考资料:
1.http://www.proteowizard.org/tools/msconvert.html
2.https://msfragger.nesvilab.org/tutorial_convert.html

samuel
你好,想请问一下.d的文件有什么开源软件来解析吗?另外,我目前在用maxquant,对.d用msconvert转换后的mgf或mzMl文件进行解析。但是遇到一些问题,似乎是bruker的这个文件格式不兼容?之前用thermo的.raw文件转换mgf或mzml格式去解析都没有问题。想请教一下.d文件转换格式的后续处理博主有推荐的pipeline吗?
陈浩
4D蛋白组学最近两年才出来,格式上软件支持不够是存在的。开源软件的话除了maxquant可以试试fragpipe.
张意一
您好,请问为啥需要选择第4步和第5步呢,这是针对timsTOF .d文件的特定设置嘛。为啥普通的数据只需要选择pickpeaking选项就行了呢
陈浩
这个示例是用timsTOF数据做演示的。因为常规的数据没有离子淌度谱( Ion mobility spectrometry,IMS )这个说法,timsTOF又称4D蛋白组学,多了一个维度。
张木心
您好,使用MSConvert转换timsTOF .d文件,右下部分,点击Add之后,表格中没有添加threshold这一行,一直是重复的scanSumming,是什么原因呢?
陈浩
我这边测试是没问题的,你用的是什么版本的MSConvert,你是选择“Threshold Peak Filter”添加后没添加上吗?建议你再试试命令行版本