除了开放的mzML、mzXML和mzData格式之外,每个供应商通常都以特定于供应商的、专有的、封闭的格式对质谱数据进行编码。为了方便我们数据分析msconvert利用厂商提供的API将这些文件转换成开放的格式。
msconvert支持的格式转换:
| Vendor | Formats | Vendor Required Software |
|---|---|---|
| ABI | T2D | DataExplorer 4.0 |
| Agilent | MassHunter .d | distributed with ProteoWizard |
| Bruker | Compass .d, YEP, BAF, FID, TDF | distributed with ProteoWizard |
| Sciex | WIFF / WIFF2 | distributed with ProteoWizard |
| Shimadzu | LCD(未完全支持) | distributed with ProteoWizard |
| Thermo Scientific | RAW | distributed with ProteoWizard |
| Waters | MassLynx .raw / UNIFI | distributed with ProteoWizard |
msconvert可转换成以下格式:
mzML 1.1
mzML 1.0
mzXML
MGF
MS2/CMS2/BMS2
mzIdentML
其中mzXML和mzML是我们常用的格式。
下载地址:
https://sourceforge.net/projects/proteowizard/
https://github.com/ProteoWizard/pwiz
备用下载:
https://pan.baidu.com/s/1fOa8c-9syk0ZbBZMvaZOIw 提取码: tsw
也可以用docker:https://hub.docker.com/r/chambm/pwiz-skyline-i-agree-to-the-vendor-licenses
安装注意事项:
Windows 用户:使用安装程序需要安装Microsoft .NET Framework 4.0或更高版本。还必须具有以下、Visual C++ redistributables组件(对于 x86 或 x64,取决于您下载的安装包版本):2008、2010、2012、2013、2015、2017。此页面链接到每个 VC 版本的最新可再发行组件,不同的供应商 DLL 依赖于不同版本的 Visual C++ redistributables组件 所以建议都安装。
示例:
1)msconvert
(输出帮助信息)
2)msconvert –help
(输出更详细的帮助信息文档)
3)msconvert data.RAW
(转换成 data.mzML到当前路径)
4)msconvert data.RAW –mzXML
(转换成 data.mzXML 到当前路径)
5)msconvert *.RAW -o my_output_dir
(转换所有以 *.RAW 为后缀的文件到 mzML并输出到 my_output_dir路径)
6)msconvert data.RAW –zlib –filter “peakPicking true [1,2]”
(用vendor方法对msLevels [1,2]进行中心化过滤,并用zlib对结果数据进行压缩,此命令比较常用)
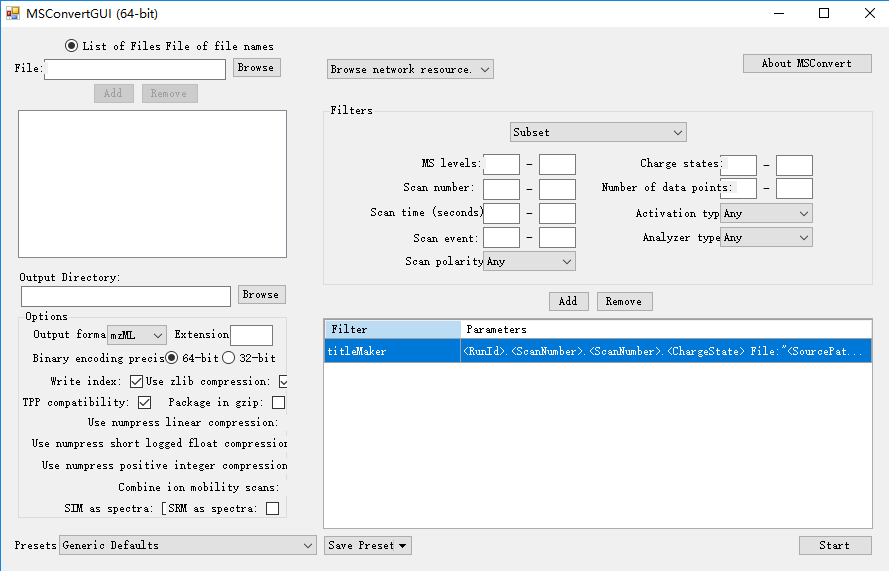
或者可以使用 ProteoWizard 自带的GUI进行可视化操作:
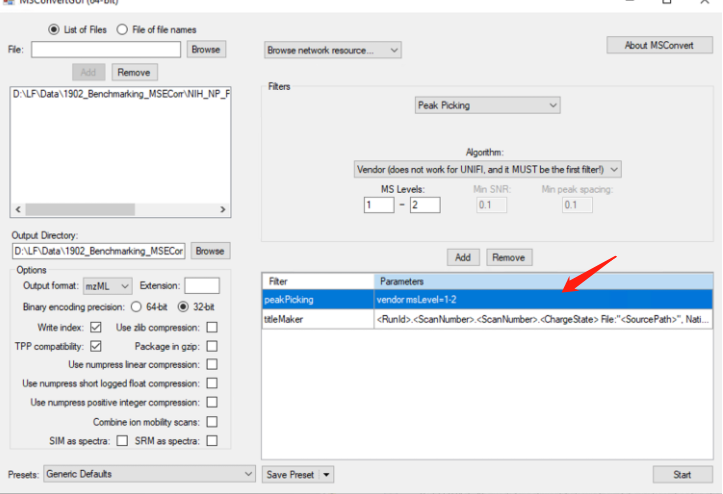
注意:当我们使用PeakPicking时,需要让其保持在第一条,否则不会进行centroided!!!
2022.04.13新增部分参数说明
最近很多来信询问设置的问题,官方文档已经有很详细的说明于是整理如下(https://proteowizard.sourceforge.io/tools/msconvert.html):
常规参数:
Options:
-f [ –filelist ] arg : specify text file containing filenames
-o [ –outdir ] arg (=.) : set output directory (‘-‘ for stdout) [.]
-c [ –config ] arg : configuration file (optionName=value)
–outfile arg : Override the name of output file.
-e [ –ext ] arg : set extension for output files
[mzML|mzXML|mgf|txt|mz5]
–mzML : write mzML format [default]
–mzXML : write mzXML format
–mz5 : write mz5 format
–mgf : write Mascot generic format
–text : write ProteoWizard internal text format
–ms1 : write MS1 format
–cms1 : write CMS1 format
–ms2 : write MS2 format
–cms2 : write CMS2 format
-v [ –verbose ] : display detailed progress information
–64 : set default binary encoding to 64-bit precision
[default]
–32 : set default binary encoding to 32-bit precision
–mz64 : encode m/z values in 64-bit precision [default]
–mz32 : encode m/z values in 32-bit precision
–inten64 : encode intensity values in 64-bit precision
–inten32 : encode intensity values in 32-bit precision
[default]
–noindex : do not write index
-i [ –contactInfo ] arg : filename for contact info
-z [ –zlib ] : use zlib compression for binary data
–numpressLinear [toler] : use numpress linear prediction lossy compression for binary mz and rt data (relative error guaranteed less than given tolerance, default is 2e-009)
–numpressPic : use numpress positive integer lossy compression for binary intensities (maximum 0.5 absolute error guaranteed)
–numpressSlof [toler] : use numpress short logged float lossy compression for binary intensities (relative error guaranteed less than given tolerance, default is 0.0002)
-n [ –numpressAll] : same as –numpressLinear –numpressSlof (see https://github.com/fickludd/ms-numpress for more info)
–numpressLinearAbsTol : desired absolute tolerance for linear numpress prediction (e.g. use 1e-4 for a mass accuracy of 0.2 ppm at 500 m/z, default uses -1.0 for maximal accuracy). Note: setting this value may substantially reduce file size, this overrides relative accuracy tolerance.
Numpress may be used at the same time as zlib (-z) for best compression, though some older mzML parsers may not handle this properly.
-g [ –gzip ] : gzip entire output file (adds .gz to filename)
–filter arg : add a spectrum list filter
–merge : create a single output file from multiple input
files by merging file-level metadata and
concatenating spectrum lists
–simAsSpectra : write selected ion monitoring as spectra, not
chromatograms
–srmAsSpectra : write selected reaction monitoring as spectra, not
chromatograms
–combineIonMobilitySpectra : write all drift bins/scans in a frame/block as one spectrum instead of individual spectra
–acceptZeroLengthSpectra : some vendor readers have an efficient way of filtering out empty spectra, but it takes more time to open the file
–ignoreUnknownInstrumentError : if true, if an instrument cannot be determined from a vendor file, it will not be an error
–help : show this message, with extra detail on filter options
这里面尤其要关注--filter参数:
index <index_value_set>
msLevel <mslevels>
chargeState <charge_states>
precursorRecalculation
mzRefiner input1.pepXML input2.mzid [msLevels=<1->] [thresholdScore=<CV_Score_Name>] [thresholdValue=<floatset>] [thresholdStep=<float>] [maxSteps=<count>]
lockmassRefiner mz=<real> mzNegIons=<real (mz)> tol=<real (1.0 Daltons)>
precursorRefine
peakPicking [<PickerType> [snr=<minimum signal-to-noise ratio>] [peakSpace=<minimum peak spacing>] [msLevel=<ms_levels>]]
scanNumber <scan_numbers>
scanEvent <scan_event_set>
scanTime <scan_time_range>
sortByScanTime
stripIT
metadataFixer
titleMaker <format_string>
threshold <type> <threshold> <orientation> [<mslevels>]
mzWindow <mzrange>
mzPrecursors <precursor_mz_list>
defaultArrayLength <peak_count_range>
zeroSamples <mode> [<MS_levels>]
mzPresent <tolerance> <type> <threshold> <orientation> <mz_list> [<include_or_exclude>]
scanSumming [precursorTol=<precursor tolerance>] [scanTimeTol=<scan time tolerance>]
MS2Denoise [<peaks_in_window> [<window_width_Da> [multicharge_fragment_relaxation]]]
MS2Deisotope [hi_res [mzTol=<mzTol>]] [Poisson [minCharge=<minCharge>] [maxCharge=<maxCharge>]]
ETDFilter [<removePrecursor> [<removeChargeReduced> [<removeNeutralLoss> [<blanketRemoval> [<matchingTolerance> ]]]]]
chargeStatePredictor [overrideExistingCharge=<true|false (false)>] [maxMultipleCharge=<int (3)>] [minMultipleCharge=<int (2)>] [singleChargeFractionTIC=<real (0.9)>] [maxKnownCharge=<int (0)>] [makeMS2=<true|false (false)>]
turbocharger [minCharge=<minCharge>] [maxCharge=<maxCharge>] [precursorsBefore=<before>] [precursorsAfter=<after>] [halfIsoWidth=<half-width of isolation window>] [defaultMinCharge=<defaultMinCharge>] [defaultMaxCharge=<defaultMaxCharge>] [useVendorPeaks=<useVendorPeaks>]
activation <precursor_activation_type>
analyzer <analyzer>
analyzerType <analyzer>
polarity <polarity>
示例如下:
# extract scan indices 5…10 and 20…25
msconvert data.RAW –filter “index [5,10] [20,25]”
# extract MS1 scans only
msconvert data.RAW –filter “msLevel 1”
# extract MS2 and MS3 scans only
msconvert data.RAW –filter “msLevel 2-3”
# extract MSn scans for n>1
msconvert data.RAW –filter “msLevel 2-“
# apply ETD precursor mass filter
msconvert data.RAW –filter ETDFilter
# remove non-flanking zero value samples
msconvert data.RAW –filter “zeroSamples removeExtra”
# remove non-flanking zero value samples in MS2 and MS3 only
msconvert data.RAW –filter “zeroSamples removeExtra 2 3”
# add missing zero value samples (with 5 flanking zeros) in MS2 and MS3 only
msconvert data.RAW –filter “zeroSamples addMissing=5 2 3”
# keep only HCD spectra from a decision tree data file
msconvert data.RAW –filter “activation HCD”
# keep the top 42 peaks or samples (depending on whether spectra are centroid or profile):
msconvert data.RAW –filter “threshold count 42 most-intense”
# multiple filters: select scan numbers and recalculate precursors
msconvert data.RAW –filter “scanNumber [500,1000]” –filter “precursorRecalculation”
# multiple filters: apply peak picking and then keep the bottom 100 peaks:
msconvert data.RAW –filter “peakPicking true 1-” –filter “threshold count 100 least-intense”
# multiple filters: apply peak picking and then keep all peaks that are at least 50% of the intensity of the base peak:
msconvert data.RAW –filter “peakPicking true 1-” –filter “threshold bpi-relative .5 most-intense
FILTER详细介绍见:https://proteowizard.sourceforge.io/tools/filters.html
参考资料:
1.http://proteowizard.sourceforge.net

不将就
你好为什么我安装的里面没有titlemaker呢,我在官网下载的
陈浩
应该都会有的,这个是在–filter参数里面进行设置的
江上渔者
您好,我想问一下,安捷伦.d文件使用MSconvert转化的MGF中MS2名称是Scan number,而直接用 MassHunter 转化的MGF中MS2是m/z。个人更倾向获得m/z,请问在MSconvert中如何设置呢?
陈浩
msconvert文档有说明,可以参考 This filter adds or replaces spectrum titles according to specified . You can use it, for example, to customize the TITLE line in MGF output in msconvert. The following keywords are recognized:
– prints the spectrum’s Run id – for example, “Data.d” from “C:/Agilent/Data.d/AcqData/mspeak.bin”
– prints the spectrum’s index
– prints the spectrum’s nativeID– prints the path of the spectrum’s source data
– if the nativeID can be represented as a single number, prints that number, else index+1
– for the first precursor, prints the spectrum’s “dissociation method” value
– for the first precursor, prints the the spectrum’s “isolation target m/z” value – prints the nativeID of the spectrum of the first precursor
– prints the m/z value of the first selected ion of the first precursor
– prints the charge state for the first selected ion of the first precursor
– prints the spectrum type
– prints the spectrum’s first scan’s start time, in seconds
– prints the spectrum’s first scan’s start time, in minutes
– prints the spectrum’s base peak m/z
– prints the spectrum’s base peak intensity
– prints the spectrum’s total ion current
– prints the spectrum’s MS level.. .“
titleMaker
For example, to create a TITLE line in msconvert MGF output with the “name.first_scan.last_scan.charge” style (eg. “mydata.145.145.2”), use –filter “titleMaker