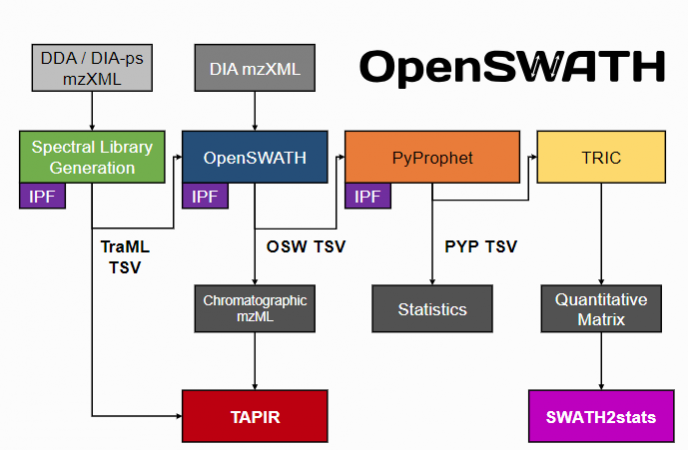
SWATH/DIA数据作为现在蛋白质组学新方式的数据 Data-Independent Acquisition (DIA) 数据,其数据分析软件相对来说以商业上Spectronaut等为主,在开源软件方面则以OpenMS等开源软件分析为主,本文以原始质谱文件出发,到最终的蛋白鉴定做一个全流程的工作流展示,旨在帮助大家批量化蛋白质组学数据分析。
需要用到的软件:Openms + Pyprophet
1)软件安装我们推荐用conda安装:
# 首先进入conda环境,前提先添加bioconda源
# 参考http://bioconda.github.io/user/install.html#set-up-channels
# conda config --add channels defaults
# conda config --add channels bioconda
# conda config --add channels conda-forge
conda install openms
conda install pyprophet2)质谱原始文件转换,需要用到msconvert,将原始质谱数据转换为公共mzML格式
详细文档请查看:http://proteowizard.sourceforge.net/tools/msconvert.html
3)准备好library文件,一般用MaxQuant建库,然后准换成OpenSWATH库的格式(此处主要介绍SWATH/DIA数据分析,不展开赘述建库流程)
OpenSWATH建库指导:http://openswath.org/en/latest/docs/generic.html
4)DIA谱图文件(mzML)搜库
for run in *.mzML
do
# 参数释义和调整请参考http://openswath.org/en/latest/docs/openswath.html
# 具体参数和所用质谱方法息息相关
OpenSwathWorkflow -in $run -tr library.pqp -tr_irt iRT_assays.TraML -swath_windows_file SWATHwindows_analysis.tsv -sort_swath_maps -batchSize 1000 -readOptions cacheWorkingInMemory -tempDirectory \Temp -use_ms1_traces -mz_extraction_window 50 -ppm -mz_correction_function quadratic_regression_delta_ppm -TransitionGroupPicker:background_subtraction original -RTNormalization:alignmentMethod linear -Scoring:stop_report_after_feature 5 -out_osw ${run}.osw
done5)搜库完成后,我们需要对搜库的结果进行鉴定打分(此处我们用pyprophet进行蛋白的鉴定和打分)
数据合并:
# 合并所有样本结果
pyprophet merge --template=library.pqp --out=merged.osw *.osw打分:
# 默认参数适用于SCIEX TripleTOF 5600/6600,其他请做适当的调整
# 如果样本量非常大,此步很慢,后续文章将会介绍高效快速的在HPC上分析
# 感兴趣的小伙伴可以看看http://openswath.org/en/latest/docs/pyprophet.html
pyprophet score --in=merged.osw --level=ms1ms2FDR计算:
# 肽水平
pyprophet peptide --context=global --in=merged.osw
# 蛋白水平
pyprophet protein --context=global --in=merged.osw导出结果:
pyprophet export --in=merged.osw --out=legacy.tsv以上是整个Workflow的全部流程,后续我们拿到肽段/蛋白矩阵就可以进行下游的分析,如:差异蛋白分析、GO/KEGG、网络、等分析。
参考资料:
1.http://openswath.org/en/latest/
