聚类是基因表达数据分析的重要工具,无论是在转录本上还是在蛋白水平上。这种无监督分类技术通常用于揭示隐藏在大型基因表达数据集中的结构。到目前为止,绝大多数应用的聚类算法都产生了数据的硬聚类,即每个基因或蛋白质被精确地分配到一个聚类中。如果集群分离良好,则硬聚类是有利的。然而,这通常不是基因表达数据的情况,因为基因/蛋白簇经常重叠。此外,硬聚类算法往往对噪声高度敏感。
在mfuzz函数中使用模糊c均值算法实现软聚类 (e1071包)
下面用一个简单示例展示Mfuzz聚类方法和效果
# 创建测试数据集
library(Mfuzz)
library(e1071)
test = matrix(rnorm(600), 200, 3)
colnames(test) = paste("Time", 1:3, sep = "")
rownames(test) = paste("Gene", 1:200, sep = "")
# 数据过滤
# NA处理
test<- filter.NA(test)
# test<- fill.NA(test)
# 生成mfuzz支持的ExpressionSet对象
df <- new("ExpressionSet", exprs = test)
# 标准化处理
df <- standardise(df)
m <- mestimate(df)
# cselection函数可以帮助确定准确的集群数量。
# 但是,应该谨慎使用,因为确定仍然很困难,特
# 别是对于短时间序列和重叠的集群。更好的方法
# 是使用一定范围的聚类数进行聚类,然后评估它
# 们的生物学相关性,例如通过GO分析。
cselection(df, m=m, crange = seq(2,5,1),repeats = 5,visu = T)
cl <- mfuzz(df, c = 6, m = m)
# 绘图
mfuzz.plot(
df,
cl,
mfrow = c(3, 2),
new.window = FALSE,
time.labels = c("Time1", "Time2", "Time3")
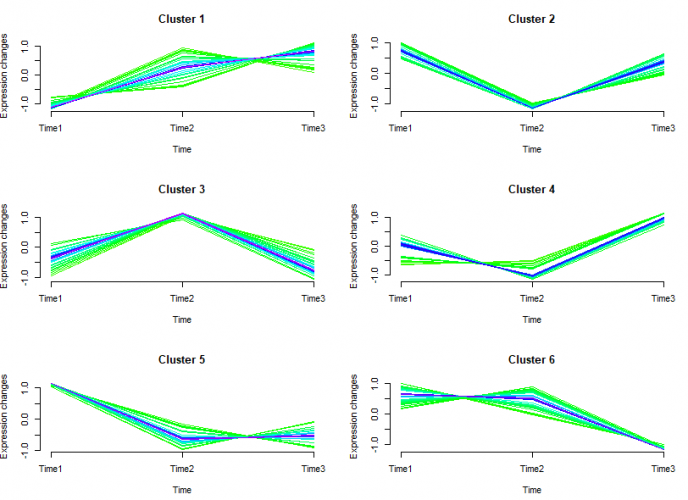
)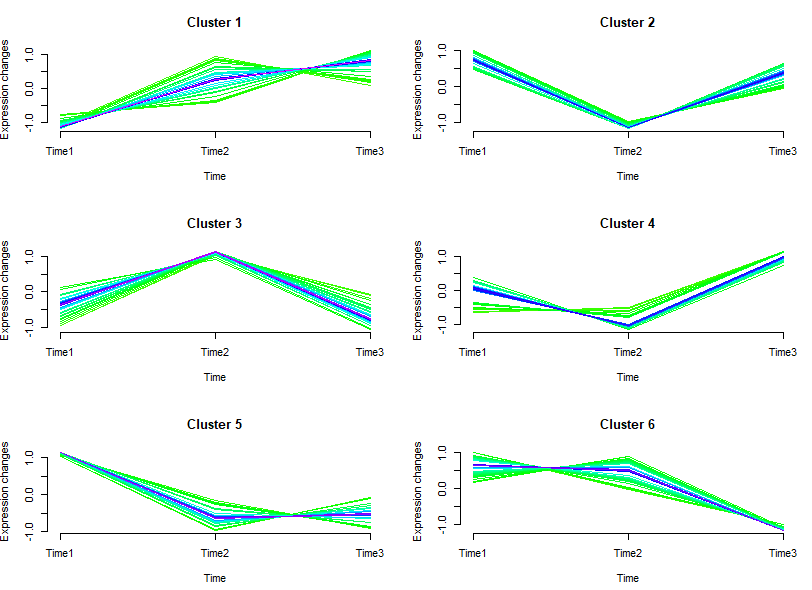
图中线条颜色越接近红色的说明是这些基因是这个cluster中的关键基因
# 导出所有基因的cluster
write.csv(cl$cluster, file = "mfuzz.csv")以上是对Mfuzz的一个简单的使用方法介绍,具体大家使用时根据自身数据集的情况来设定参数使用。
参考资料:
1.https://blog.csdn.net/zjsghww/article/details/50922168
2.http://www.bioconductor.org/packages/release/bioc/html/Mfuzz.html

12345
请问这个图可以加上图例吗?
陈浩
是要添加什么样的图例呢?这个趋势就是时间点的图
dot
您好,请问mfuzz每次的结果不一致是不是因为数据不适合用c means聚类?
陈浩
不是的,你仔细观察比如第一次图的Cluster1可能第二次的某个图很相近或者一样;但是如果你仔细观察会发现少许的不一致,这个是因为cmeans进行迭代,每次起始的迭代是随机开始点的,另外迭代次数不一样结果也会有些许不一致,所以不会产生一模一样的图,但是大致趋势会一样的。