聚类是基因表达数据分析的重要工具,无论是在转录本上还是在蛋白水平上。这种无监督分类技术通常用于揭示隐藏在大型基因表达数据集中的结构。到目前为止,绝大多数应用的聚类算法都产生了数据的硬聚类,即每个基因或蛋白质被精确地分配到一个聚类中。如果集群分离良好,则硬聚类是有利的。然而,这通常不是基因表达数据的情况,因为基因/蛋白簇经常重叠。此外,硬聚类算法往往对噪声高度敏感。
在mfuzz函数中使用模糊c均值算法实现软聚类 (e1071包)
下面用一个简单示例展示Mfuzz聚类方法和效果
# 创建测试数据集
library(Mfuzz)
library(e1071)
test = matrix(rnorm(600), 200, 3)
colnames(test) = paste("Time", 1:3, sep = "")
rownames(test) = paste("Gene", 1:200, sep = "")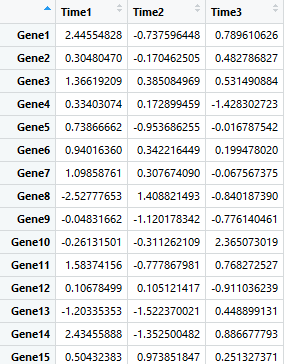
# 数据过滤
# NA处理
test<- filter.NA(test)
# test<- fill.NA(test)
# 生成mfuzz支持的ExpressionSet对象
df <- new("ExpressionSet", exprs = test)
# 标准化处理
df <- standardise(df)
m <- mestimate(df)
# cselection函数可以帮助确定准确的集群数量。
# 但是,应该谨慎使用,因为确定仍然很困难,特
# 别是对于短时间序列和重叠的集群。更好的方法
# 是使用一定范围的聚类数进行聚类,然后评估它
# 们的生物学相关性,例如通过GO分析。
cselection(df, m=m, crange = seq(2,5,1),repeats = 5,visu = T)
cl <- mfuzz(df, c = 6, m = m)
# 绘图
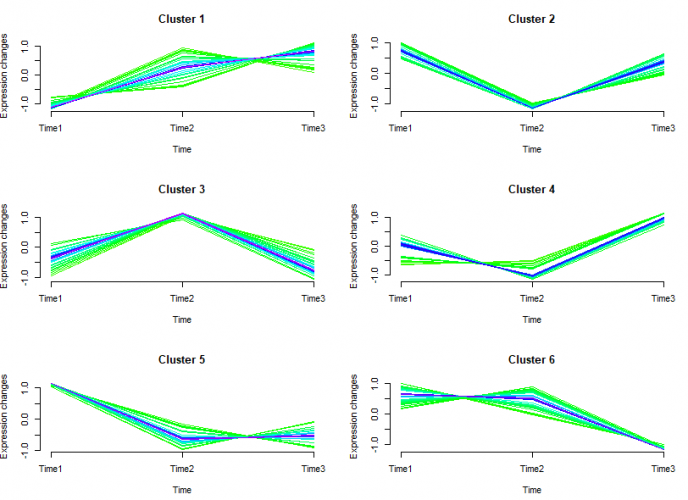
mfuzz.plot(
df,
cl,
mfrow = c(3, 2),
new.window = FALSE,
time.labels = c("Time1", "Time2", "Time3")
)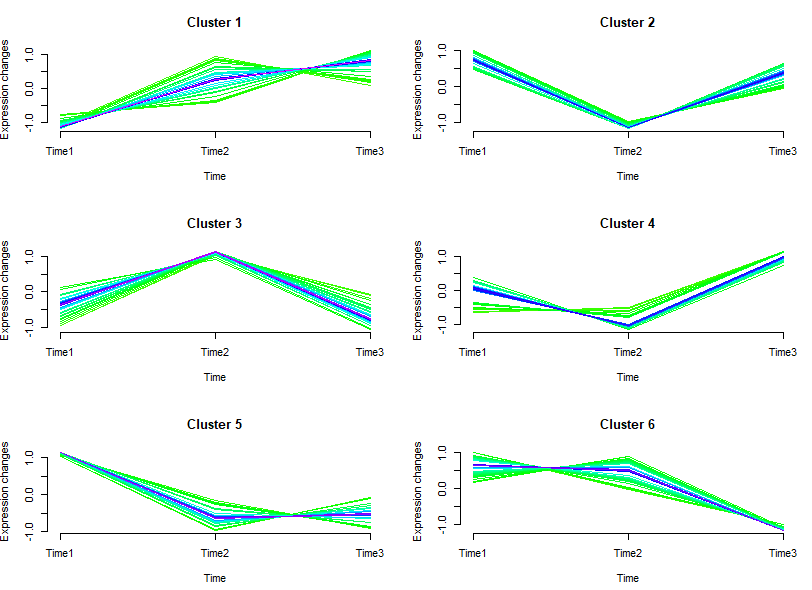
图中线条颜色越接近红色的说明是这些基因是这个cluster中的关键基因
# 导出所有基因的cluster
write.csv(cl$cluster, file = "mfuzz.csv")以上是对Mfuzz的一个简单的使用方法介绍,具体大家使用时根据自身数据集的情况来设定参数使用。
参考资料:
1.https://blog.csdn.net/zjsghww/article/details/50922168
2.http://www.bioconductor.org/packages/release/bioc/html/Mfuzz.html

秒秒
您好,同请问Mfuzz两次运行结果为什么不一样?是用一样的代码跑了两遍
夏希
Mfuzz相同结果相同处理,为什么两次出来的结果不一样,也就是重复性不好是因为软聚类引起的吗?
陈浩
一般不会因为是聚类引起的,mfuzz采用的是e1071包的cmeans函数进行聚类的。猜测的可能是cluster的顺序不一样,你可以将你的代码或者测试数据发至我的邮箱,我帮你看看(邮箱地址见页面最下方或者关于页面联系我)。
夏希
你好,关于我的代码和数据我已经发您邮箱
陈浩
已答复
秒秒
您好,同请问Mfuzz两次运行结果为什么不一样呢?是用一样代码跑了两遍
陈浩
已经邮件回复
小童
您好,请问Mfuzz 的哪个输出文件,可以后续使用ggplot2 进行可视化呢
陈浩
Mfuzz输出的主要是聚类cluster1~N的信息,这个用于后续的绘制折线图等,可以参考下我的这篇文章:https://evvail.com/2020/08/03/1076.html,希望能帮到你。
陈小年
您好,我想请教一个问题:如何理解绘图的结果中的纵坐标expression changes?我有两点困惑,1)如果是类似于差异基因foldchange,那么这里是谁跟谁比出来的变化倍数?2)如果是类似于表达量scale,那么为什么每个gene cluster的变化范围都一样都是-1,1?我的数据是-1.5,1.5。盼复,谢谢!
陈浩
你好,这是一个很好的问题,
1)这里的数据表达值不是谁和谁比,是对每一个基因做了一个软聚类,每一个横坐标是一个时间点或者状态,纵坐标是进行了归一化/标准化的表达值,Mfuzz主要是对其做了一个软聚类,可以理解为把有相同趋势的基因分在同一个cluster里面了
2)-1,1是做了归一化,这个根据你的数据具体对待
希望这个解释能帮到你。
陈小年
好的谢谢回复,我理解了。expression changes相当于做差异基因表达量热图的时候的归一化结果z-score,我的数据高表达和低表达量差的比较多就是-1.5,1.5.
Shichao Sun
您好,用Mfuzz软件直接出的图片达不到发文章要求,能改些参数对其美化一下吗?
陈浩
可以的,Mfuzz可以导出其数据用ggplot2等绘制的。
李志奇
信件已发,请您有空的时候看一下,谢谢~
陈浩
好的,晚上我看一下回复你。
李志奇
我是想对模块基因进行一个可视化,如果用ggplot2画的话,我这边导出的是模块对应基因的表达量RPKM值,用ggplot2画的话纵坐标应该用什么值来表示呢?
陈浩
1)如果你是看每个模块间的基因的关系,推荐热图(pheatmap),行是基因,列是样本(列标签moudel+样本分类)值可以是RPKM也可以是权重矩阵或者网络图(用cytoscape展示,用exportNetworkToCytoscape导出)
2)如果你是看每个模块间的关系WGCNA自带的TOMplot这个就挺好的,行列均是基因;
3)如果是关心具体某个模块内部的基因:i.看模块与样本分类之间的相关性,可以绘制热图(行是基因,列是样本),也可以绘制散点图(算出每个模块跟基因的相关系数矩阵,看模块和样本分类的一个相关性);ii.看模块内部基因在不同的样本类型中的一个表达形况,可以绘制热图,同1。
根据不同的场景来选择,WGCNA相关的分析常用的展示方式就是热图、网络图、散点/折线图。
李志奇
非常感谢您的回答,请问我如何用模块基因绘制表达模式折线图的话,纵坐标可以用每个基因的表达平均值,然后各个样品跟他进行相除,然后换算log2后的值吗,来绘制这个表达模式折线图
陈浩
1)RPKM是已经标准化后的值不需要再次进行log
2)该基因表达值除以均值只是范围被缩放了,对图没有实际影响
可以y轴用基因,x轴用样本表达值绘制,类似
 一样。
一样。
李志奇
请问可以加一下您的QQ吗,我这边好像没法把我画的图的图片放上来。
陈浩
抱歉,我现在不用QQ。你可以发到我的邮箱chenhao(at)evvail.com!
李志奇
我的想法是用每个模块分别做一个模式图,最后进行图片拼接,但是这边的C设置为1,进行聚类就会报错,代码如下。
如何决定聚类个数?
> c # 评估出最佳的m值
> m # 聚类
> cl <- mfuzz(eset,c=c,m=m)
Error in apply(u, 1, which.max) : dim(X) must have a positive length,请问有什么解决的办法呢?
陈浩
mfuzz是用了e1071包的C-means的一种聚类算法,cluster数需要大于1(其中源码中“cluster <- apply(u, 1, which.max)”,这个需要指定的cluster大于1)。你要把每个基因都显示,我个人建议热图或者折线图是最直接的。你用mfuzz的目的如果只是需要画一个折线图,建议用ggplot2,如果你需要用到聚类的话,可以按照我之前建议的思路。
李志奇
我现在已经把分类模块以及对应的基因表达量导出来了,如果用计算模块基因平均值好像不能把每个基因的趋势都在图上边显示出来吧,请问可视化基因表达模式趋势的画还有什么别的包可以画呢,对于已经用WGCNA模块分类出来的结果
陈浩
是的,前面我描述过。其实WGCNA热图还是挺直观的,你可以定制下热图,参考:https://evvail.com/2020/05/21/546.html。如果你想用其他形式把每个基因的趋势都画出来,建议你画每个model的基因折线图或者山峦图,这个在我的博客也有详细的描述和Code你可以参考。
李志奇
请问Mfuzz可以对已经聚类好的模块进行可视化吗,我想对WGCNA模块分类结果进行基因表达趋势图的绘制
陈浩
一般WGCNA寻找到的module都属于相关性较高的基因,其实可以通过官方自带的热图也能说明各个模块的一个趋势,直接用Mfuzz对基因表达值聚类方法不太可行,因为一个基因理论上属于一个module,表达值只存在一个module中,所以通过基因的方式行不通。
但是如果你是要通过样本表达值对module的趋势来做,理论上是ok的。如果你这边想用Mfuzz这种软聚类的方式对各个module的趋势做一个研究思路上是可行的,假设你的数据是这样的,并且你已经提取module的基因和相应的值:
model sample_1 sample_2 sample_3 sample_4 sample_5 sample_6 sample_7 sample_8 sample_9
gene1 module1 47 39 69 84 37 21 73 98 87
gene2 module1 85 87 86 42 32 57 73 57 47
gene3 module1 89 43 91 32 50 40 6 91 61
gene4 module1 65 59 77 70 42 35 19 4 21
gene5 module1 14 59 7 87 66 5 10 33 24
gene6 module1 98 42 95 76 50 74 51 91 74
gene7 module1 87 70 19 12 49 53 93 72 14
gene8 module1 75 12 18 48 94 61 93 100 22
gene9 module1 26 60 82 25 14 85 98 63 19
gene10 module2 83 78 7 69 94 74 4 45 84
gene11 module2 92 40 79 95 91 96 14 54 58
gene12 module2 21 37 91 50 1 68 39 27 19
gene13 module2 16 79 93 11 38 36 3 28 23
gene14 module2 19 31 16 55 96 20 60 83 20
gene15 module2 97 31 51 61 11 56 18 54 63
gene16 module3 90 27 81 53 52 26 99 61 53
gene17 module3 29 14 23 58 78 63 13 39 25
gene18 module3 19 92 27 94 15 67 32 62 10
gene19 module3 86 9 54 71 74 27 27 79 89
gene20 module4 6 40 64 59 93 7 13 9 21
gene21 module4 56 21 51 8 92 43 37 73 52
gene22 module4 69 72 25 18 38 69 91 40 78
gene23 module4 27 71 8 96 56 12 33 51 17
gene24 module4 86 47 31 25 4 86 8 100 9
gene25 module4 65 50 32 15 36 20 24 47 65
gene26 module4 62 2 4 92 90 64 75 39 44
gene27 module4 60 30 3 32 73 57 33 93 82
gene28 module4 13 59 21 81 62 41 18 38 82
这样你首先需要构造出Mfuzz需要的矩阵,这里面最主要的是对module聚类的这个思想,因为WGCNA已经对GENE做了关键模块的挖掘,所以现在要做的是看各个模块的一个趋势,首先计算各个样本的基因再各个模块下的均值(或者中位数等,module都属于相关性较高的基因,此处用均值相对来说推荐)
module1 module2 module3 module4
sample_1 9 81 20 97
sample_2 100 47 97 42
sample_3 69 40 40 5
sample_4 30 16 6 88
sample_5 84 59 7 89
sample_6 53 98 35 85
sample_7 55 17 20 12
sample_8 59 57 75 60
sample_9 89 88 79 67
绘图其实相对很简单了。这里面需要注意的点就是这里面哪一个module作为起始点,以及什么样的module趋势是你研究需要的,这个可能要根据你的课题做出假设了。我们假设module1~4顺序排布,就构造上面的矩阵,剩下的就和常规的一致了,这样挖掘出来的趋势可能要结合你研究的特点,如果某一个cluster能恰好把你不同的moudle区分开,那应该是一篇大作了。
希望能帮到你!