可变剪切是指mRNA前体可以有多种方式将exon连接在一起的过程。 由于可变剪切使一个基因产生多个转录本,不同转录本可能翻译成不同蛋白。在传统的转录组未考虑可变剪接的存在,这意味着RNA-seq数据的全部信息通常利用不充分。
IsoformSwitchAnalyzeR它能够根据RNA序列衍生的新型和/或带注释的全长同工型的定量数据对同工型转换进行统计鉴定。IsoformSwitchAnalyzeR有助于集成(预测的)注释的许多来源,例如开放阅读框(ORF / CDS),蛋白结构域(通过Pfam),信号肽(通过SignalP),无固定三维结构的蛋白(IDR,通过NetSurfP-2或IUPred2A),编码潜力(通过CPAT或CPC2)以及转录调控机制(NMD)的敏感性等。
总之,IsoformSwitchAnalyzeR能够更细致的分析RNA-seq数据,重点是同工型转换(具有预期的结果)及其相关的可变剪接,从而扩大了RNA-seq数据的可用性。
1)安装
if (!requireNamespace("BiocManager", quietly = TRUE)){
install.packages("BiocManager")
BiocManager::install("IsoformSwitchAnalyzeR")
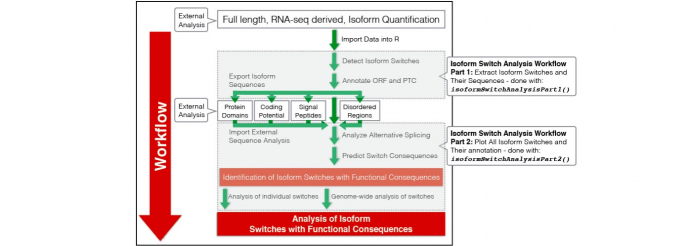
}2)Workflow
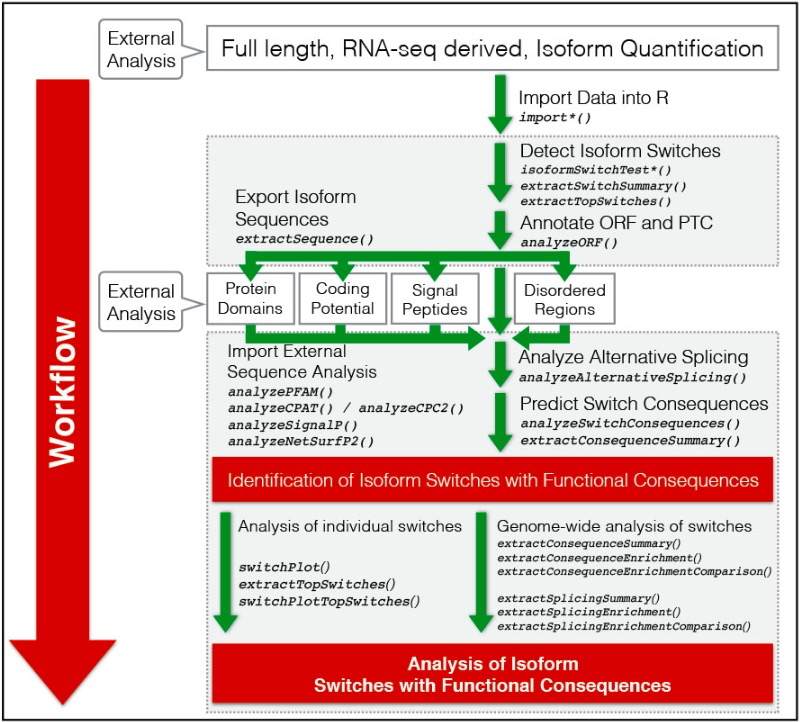
3)一个简单的示例
library(IsoformSwitchAnalyzeR)
# 导入salmon数据
salmonQuant <- importIsoformExpression(
parentDir = system.file("extdata/",package="IsoformSwitchAnalyzeR")
)
# 试验设计
myDesign <- data.frame(
sampleID = colnames(salmonQuant$abundance)[-1],
condition = gsub('_.*', '', colnames(salmonQuant$abundance)[-1])
)
# 构建 switchAnalyzeRlist 对象
aSwitchList <- importRdata(
isoformCountMatrix = salmonQuant$counts,
isoformRepExpression = salmonQuant$abundance,
designMatrix = myDesign,
isoformExonAnnoation = system.file("extdata/example.gtf.gz" , package="IsoformSwitchAnalyzeR"),
# 转录本的 fasta 序列文件
isoformNtFasta = system.file("extdata/example_isoform_nt.fasta.gz", package="IsoformSwitchAnalyzeR"),
fixStringTieAnnotationProblem = TRUE,
showProgress = FALSE
)
# 过滤
aSwitchList <- preFilter(aSwitchList)
# 分析差异表达的isoform
aSwitchListAnalyzed <- isoformSwitchTestDEXSeq(
switchAnalyzeRlist = aSwitchList,
reduceToSwitchingGenes=TRUE
)
# OFR分析
exampleSwitchListAnalyzed <- analyzeORF(
aSwitchListAnalyzed,
orfMethod = "longest",
showProgress=FALSE
)
exampleSwitchListAnalyzed <- extractSequence(
exampleSwitchListAnalyzed,
pathToOutput = '<insert_path>',
writeToFile=FALSE
)
# 还有很多分析,如:
# analyzeCPAT() # OR
# analyzeCPC2()
# analyzePFAM()
# analyzeSignalP()
# analyzeIUPred2A() # OR
# analyzeNetSurfP2()
### Add CPC2 analysis
exampleSwitchListAnalyzed <- analyzeCPC2(
switchAnalyzeRlist = exampleSwitchListAnalyzed,
pathToCPC2resultFile = system.file("extdata/cpc2_result.txt", package = "IsoformSwitchAnalyzeR"),
removeNoncodinORFs = TRUE # because ORF was predicted de novo
)
#> Added coding potential to 162 (100%) transcripts
### Add PFAM analysis
exampleSwitchListAnalyzed <- analyzePFAM(
switchAnalyzeRlist = exampleSwitchListAnalyzed,
pathToPFAMresultFile = system.file("extdata/pfam_results.txt", package = "IsoformSwitchAnalyzeR"),
showProgress=FALSE
)
#> Converting AA coordinats to transcript and genomic coordinats...
#> Added domain information to 127 (78.4%) transcripts
### Add SignalP analysis
exampleSwitchListAnalyzed <- analyzeSignalP(
switchAnalyzeRlist = exampleSwitchListAnalyzed,
pathToSignalPresultFile = system.file("extdata/signalP_results.txt", package = "IsoformSwitchAnalyzeR")
)
#> Added signal peptide information to 17 (10.49%) transcripts
### Add NetSurfP2 analysis
exampleSwitchListAnalyzed <- analyzeIUPred2A(
switchAnalyzeRlist = exampleSwitchListAnalyzed,
pathToIUPred2AresultFile = system.file("extdata/iupred2a_result.txt.gz", package = "IsoformSwitchAnalyzeR"),
showProgress = FALSE
)
# 进行可变剪切分析
exampleSwitchListAnalyzed <- analyzeAlternativeSplicing(
switchAnalyzeRlist = exampleSwitchListAnalyzed,
quiet=TRUE
)
# 提取异构体中其异构体使用量有显着变化和异构体的异构体
consequencesOfInterest <- c('intron_retention','coding_potential','NMD_status','domains_identified','ORF_seq_similarity')
exampleSwitchListAnalyzed <- analyzeSwitchConsequences(
exampleSwitchListAnalyzed,
consequencesToAnalyze = consequencesOfInterest,
dIFcutoff = 0.4, # very high cutoff for fast runtimes - you should use the default (0.1)
showProgress=FALSE
)
# 异构体分析
exampleSwitchListAnalyzedSubset <- subsetSwitchAnalyzeRlist(
exampleSwitchListAnalyzed,
exampleSwitchListAnalyzed$isoformFeatures$condition_1 == 'COAD_ctrl'
)
# 绘图
switchPlot(exampleSwitchListAnalyzedSubset, gene = 'ZAK')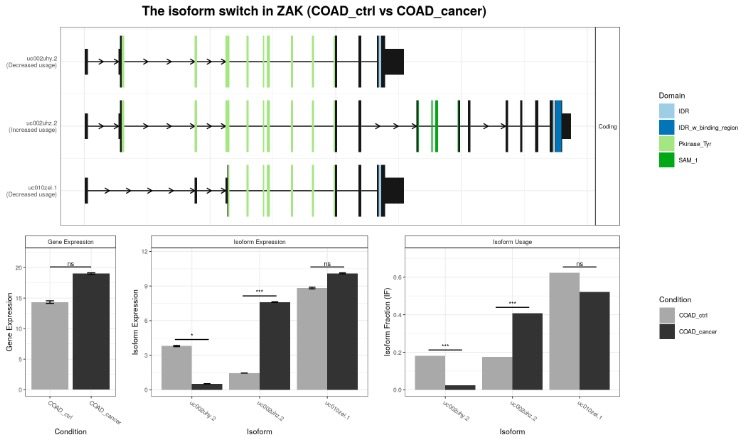
也可以单独的绘制每个图:
switchPlotTranscript(exampleSwitchListAnalyzedSubset, gene = 'ZAK')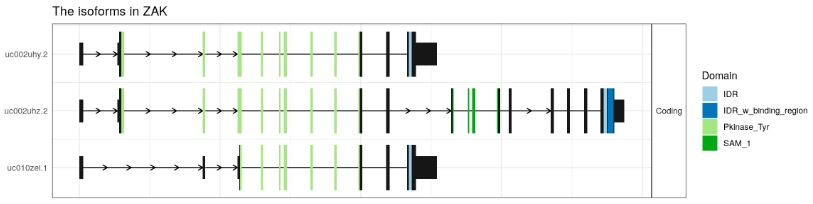
switchPlotGeneExp (exampleSwitchListAnalyzedSubset, gene = 'ZAK')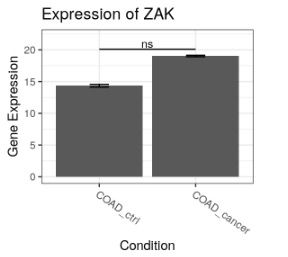
switchPlotIsoExp(exampleSwitchListAnalyzedSubset, gene = 'TNFRSF1B')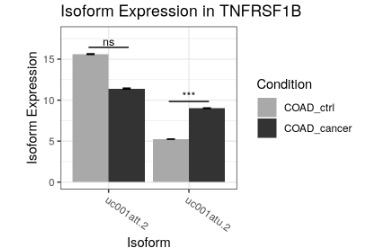
switchPlotIsoUsage(exampleSwitchListAnalyzedSubset, gene = 'ZAK')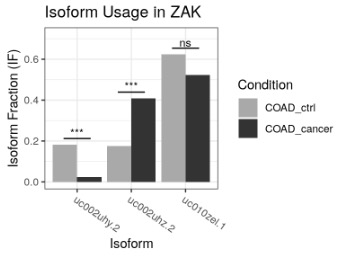
IsoformSwitchAnalyzeR该包含了诸多的分析内容,是转录本分析的一大神器,推荐阅读官方文档进行全面了解和学习。
4)IsoformSwitchAnalyzeR中用到的一些方法及其参考文献/资料
- Import of data from Salmon/Kallisto/RSEM/StringTie (importRdata() function): Please cite reference 10.
- Import of data from Salmon via Tximeta (importSalmonData() function): Please cite reference 10 and 17.
- Inter-library normalization of abundance values: Please cite reference 10 and 11.
- Isoform switch test implemented utilizing DEXSeq via IsoformSwitchAnalyzeR (Default) : Please cite reference 1, 12 and 13.
- Isoform switch test implemented in the DRIMSeq package: Please cite reference 1 and 3.
- Prediction of open reading frames (ORF) analysis: Please cite reference 1 and 4.
- Prediction of pre-mature termination codons (PTC) and thereby NMD-sensitivity: Please cite reference 1, 4, 5 and 6.
- CPAT: Please cite reference 7.
- CPC2: Please cite reference 14.
- Pfam: Please cite reference 8.
- SignalP: Please cite reference 9.
- NetSurf2-P: Please cite reference 15.
- IUPred2A: Please cite reference 16.
- Prediction of consequences: Please cite reference 1.
- Visualizations (plots) implemented in the IsoformSwitchAnalyzeR package: Please cite reference 1.
- Alternative splicing analysis: Please cite both reference 1 and 4.
- Genome-wide enrichment analysis: Please cite both reference 1 and 2.
Refrences:
- Vitting-Seerup et al. The Landscape of Isoform Switches in Human Cancers. Cancer Res. (2017) link.
- Vitting-Seerup et al. IsoformSwitchAnalyzeR: Analysis of changes in genome-wide patterns of alternative splicing and its functional consequences. Bioinformatics (2019) link.
- Nowicka et al. DRIMSeq: a Dirichlet-multinomial framework for multivariate count outcomes in genomics. F1000Research, 5(0), 1356. link.
- Vitting-Seerup et al. spliceR: an R package for classification of alternative splicing and prediction of coding potential from RNA-seq data. BMC Bioinformatics 2014, 15:81. link.
- Weischenfeldt et al. Mammalian tissues defective in nonsense-mediated mRNA decay display highly aberrant splicing patterns. Genome Biol 2012, 13:R35 link.
- Huber et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods, 2015, 12:115-121. link.
- Wang et al. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41:e74. link.
- Finn et al. The Pfam protein families database. Nucleic Acids Research (2012) link.
- _Almagro et al. SignalP 5.0 improves signal peptide predictions using deep neural networks.**. Nat. Biotechnol (2019)_ link
- Soneson et al. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research 4, 1521 (2015). link.
- Robinson et al. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biology (2010) link.
- Ritchie et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research (2015) link.
- Anders et al. Detecting differential usage of exons from RNA-seq data. Genome Research (2012) link.
- Kang et al. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res (2017) link.
- Klausen et al. NetSurfP-2.0: improved prediction of protein structural features by integrated deep learning. BioRxiv (2018) link
- Meszaros et al. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res (2018) link
- Love et al. Tximeta: Reference sequence checksums for provenance identification in RNA-seq. PLoS Comput. Biol (2020) link
参考资料:
1.http://bioconductor.org/packages/release/bioc/vignettes/IsoformSwitchAnalyzeR/inst/doc/IsoformSwitchAnalyzeR.html#importing-data-from-salmon-via-tximeta
