CancerSubtypes是用于癌症亚型分析的R包,其中包括了从数据集处理到结果验证的各种功能。其主要功能包括基因组数据预处理,癌症亚型鉴定,结果验证,可视化和差异比较。
CancerSubtypes为基因组数据预处理提供了常见的数据插补和归一化方法。同时包含了常见的癌症亚型识别方法(Consensus clustering (CC) 、Consensus Nonnegative matrix factorization (CNMF)、Similarity Network Fusion (SNF)、Combined SNF and CC (SNF.CC)、Weighted Similarity Network Fusion (WSNF))
下面安装CancerSubtypes:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("CancerSubtypes")1)数据处理,CancerSubtypes包中有四种特征选择方法(方差-Var,中位数绝对偏差-MAD,COX模型,主成分分析-PCA)
### Prepare a TCGA gene expression dataset for analysis.
library(CancerSubtypes)
library("RTCGA.mRNA")
rm(list = ls())
data(BRCA.mRNA)
mRNA=t(as.matrix(BRCA.mRNA[,-1]))
colnames(mRNA)=BRCA.mRNA[,1]
###To observe the mean, variance and Median Absolute Deviation distribution of the dataset, it helps users to get the distribution characteristics of the data, e.g. To evaluate whether the dataset fits a normal distribution or not.
data.checkDistribution(mRNA)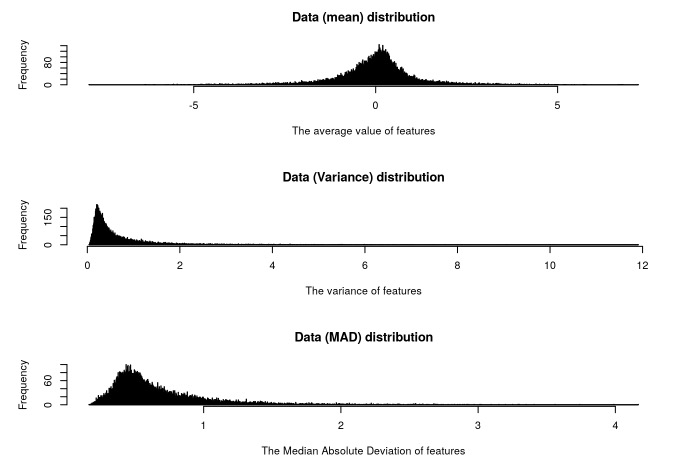
缺失值计算:
# CancerSubtypes整合了三种常见的基因组数据插补方法
index=which(is.na(mRNA))
res1=data.imputation(mRNA,fun="median")
res2=data.imputation(mRNA,fun="mean")
res3=data.imputation(mRNA,fun="microarray")数据标准化:
result1=data.normalization(mRNA,type="feature_Median",log2=FALSE)
result2=data.normalization(mRNA,type="feature_zscore",log2=FALSE)2)特征选择
# 基于最大方差的特征选择
###The top 1000 most variance features will be selected.
data1=FSbyVar(mRNA, cut.type="topk",value=1000)
###The features with (variance>0.5) are selected.
data2=FSbyVar(mRNA, cut.type="cutoff",value=0.5)
# 基于变化最大的中位数绝对偏差(MAD)进行特征选择
data1=FSbyMAD(mRNA, cut.type="topk",value=1000)
data2=FSbyMAD(mRNA, cut.type="cutoff",value=0.5)
# 基于主成分分析的特征维约简和提取
mRNA1=data.imputation(mRNA,fun="microarray")
data1=FSbyPCA(mRNA1, PC_percent=0.9,scale = TRUE)
# 基于Cox回归模型的特征选择
data(GeneExp)
data(time)
data(status)
data1=FSbyCox(GeneExp,time,status,cutoff=0.05)3)癌症亚型鉴定
Consensus clustering (CC, 2003) 是一种无监督的亚型发现方法,是许多基因组学研究中经常使用且有价值的方法,并具有许多成功的应用
### The input dataset is single gene expression matrix.
data(GeneExp)
result=ExecuteCC(clusterNum=3,d=GeneExp,maxK=10,clusterAlg="hc",distance="pearson",title="GBM")
### The input dataset is multi-genomics data as a list
data(GeneExp)
data(miRNAExp)
GBM=list(GeneExp=GeneExp,miRNAExp=miRNAExp)
result=ExecuteCC(clusterNum=3,d=GBM,maxK=10,clusterAlg="hc",distance="pearson",title="GBM")Non-negative matrix factorization 非负矩阵分解(CNMF,2004)作为一种有效的降维方法,被用于区分高维基因组数据的分子模式,并为分类发现提供了一种有力的方法。我们应用NMF包对癌症基因组数据集执行非负矩阵分解。
### The input dataset is single gene expression matrix.
data(GeneExp)
result=ExecuteCNMF(GeneExp,clusterNum=3,nrun=30)
### The input dataset is multi-genomics data as a list
data(GeneExp)
data(miRNAExp)
GBM=list(GeneExp=GeneExp,miRNAExp=miRNAExp)
result=ExecuteCNMF(GBM,clusterNum=3,nrun=30)Integrative clustering (iCluster, 2009) 使用联合潜变量模型对多种组学数据进行集成聚类。
data(GeneExp)
data(miRNAExp)
data1=FSbyVar(GeneExp, cut.type="topk",value=1000)
data2=FSbyVar(miRNAExp, cut.type="topk",value=300)
GBM=list(GeneExp=data1,miRNAExp=data2)
result=ExecuteiCluster(datasets=GBM, k=3, lambda=list(0.44,0.33,0.28))Similarity network fusion 相似性网络融合(SNF,2014)是融合相似性网络的一种计算方法,用于聚合多组学数据。
data(GeneExp)
data(miRNAExp)
GBM=list(GeneExp=GeneExp,miRNAExp=miRNAExp)
result=ExecuteSNF(GBM, clusterNum=3, K=20, alpha=0.5, t=20)Combined SNF and CC (SNF.CC)将SNF和CC结合在一起以产生一种新的癌症亚型鉴定方法
data(GeneExp)
data(miRNAExp)
data(time)
data(status)
data1=FSbyCox(GeneExp,time,status,cutoff=0.05)
data2=FSbyCox(miRNAExp,time,status,cutoff=0.05)
GBM=list(GeneExp=data1,miRNAExp=data2)
result=ExecuteSNF.CC(GBM, clusterNum=3, K=20, alpha=0.5, t=20,maxK = 10, pItem = 0.8,reps=500,
title = "GBM", plot = "png", finalLinkage ="average")Weighted Similarity network fusion(WSNF)是在基因调控网络信息的帮助下进行的一种癌亚型鉴定方法。它利用miRNA-TF-mRNA调控网络来考虑功能的重要性。
data(GeneExp)
data(miRNAExp)
data(Ranking)
####Retrieve there feature ranking for genes
gene_Name=rownames(GeneExp)
index1=match(gene_Name,Ranking$mRNA_TF_miRNA.v21_SYMBOL)
gene_ranking=data.frame(gene_Name,Ranking[index1,],stringsAsFactors=FALSE)
index2=which(is.na(gene_ranking$ranking_default))
gene_ranking$ranking_default[index2]=min(gene_ranking$ranking_default,na.rm =TRUE)
####Retrieve there feature ranking for genes
miRNA_ID=rownames(miRNAExp)
index3=match(miRNA_ID,Ranking$mRNA_TF_miRNA_ID)
miRNA_ranking=data.frame(miRNA_ID,Ranking[index3,],stringsAsFactors=FALSE)
index4=which(is.na(miRNA_ranking$ranking_default))
miRNA_ranking$ranking_default[index4]=min(miRNA_ranking$ranking_default,na.rm =TRUE)
###Clustering
ranking1=list(gene_ranking$ranking_default ,miRNA_ranking$ranking_default)
GBM=list(GeneExp,miRNAExp)
result=ExecuteWSNF(datasets=GBM, feature_ranking=ranking1, beta = 0.8, clusterNum=3,
K = 20,alpha = 0.5, t = 20, plot = TRUE)4)结果验证及可视化
轮廓宽度用于测量与其他子类型相比,样本与其标识的子类型匹配的相似程度,高值表示样本匹配良好。每条水平线代表轮廓图中的一个样本,线的长度是样本具有的轮廓宽度。
data(GeneExp)
data(miRNAExp)
GBM=list(GeneExp=GeneExp,miRNAExp=miRNAExp)
result=ExecuteSNF(GBM, clusterNum=3, K=20, alpha=0.5, t=20,plot = FALSE)
###Similarity smaple matrix
sil=silhouette_SimilarityMatrix(result$group, result$distanceMatrix)
plot(sil)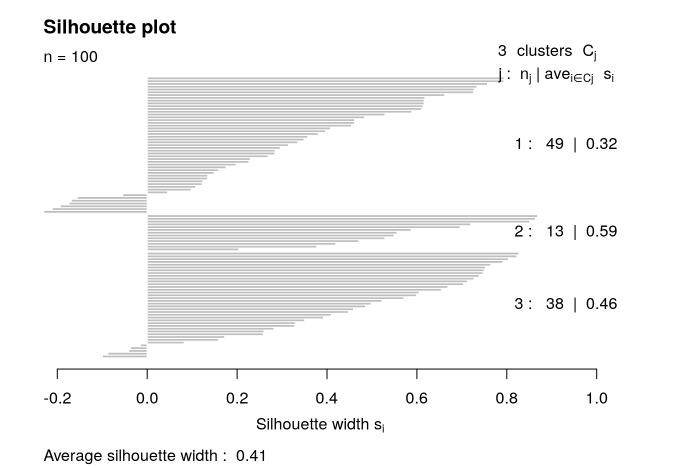
生存分析用于判断亚型之间的不同生存模式。
data(GeneExp)
data(miRNAExp)
data(time)
data(status)
data1=FSbyCox(GeneExp,time,status,cutoff=0.05)
data2=FSbyCox(miRNAExp,time,status,cutoff=0.05)
GBM=list(GeneExp=data1,miRNAExp=data2)
#### 1.ExecuteSNF
result1=ExecuteSNF(GBM, clusterNum=3, K=20, alpha=0.5, t=20,plot = FALSE)
group1=result1$group
distanceMatrix1=result1$distanceMatrix
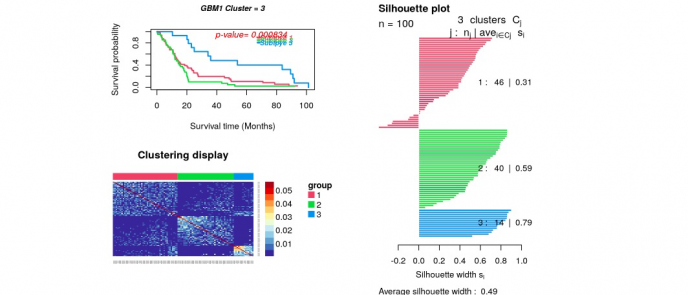
p_value=survAnalysis(mainTitle="GBM1",time,status,group1,
distanceMatrix1,similarity=TRUE)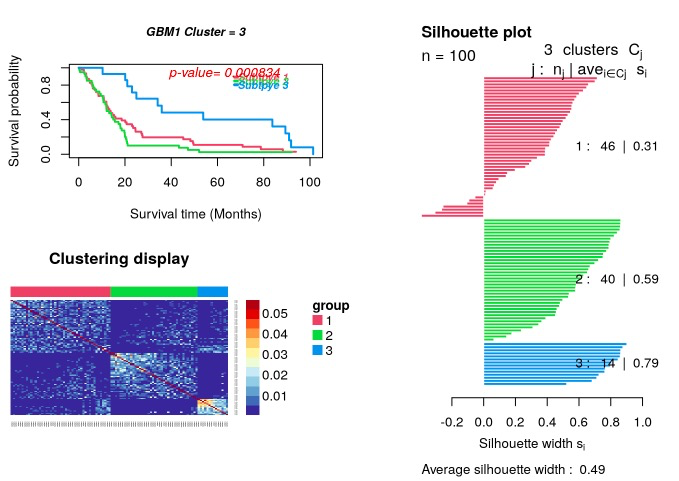
聚类的统计显着性是一种纯统计方法,用于检验子类型之间的显着性差异数据分布。不同的表达是为了检验每种亚型与参考组(对照样品/正常样品)之间的表达差异。
data(GeneExp)
data(miRNAExp)
data(time)
data(status)
GBM=list(GeneExp=GeneExp,miRNAExp=miRNAExp)
result=ExecuteSNF(GBM, clusterNum=3, K=20, alpha=0.5, t=20,plot = FALSE)
group=result$group
sigclust=sigclustTest(miRNAExp,group, nsim=1000, nrep=1, icovest=1)
sigclust差异表达分析是为了检验每个亚型与参考组(对照样品/正常样品)之间的表达差异。CancerSubtypes用limma软件包对每个亚型和正常样本之间进行不同的表达分析。
library("RTCGA.mRNA")
#require(TCGAbiolinks)
rm(list = ls())
data(BRCA.mRNA)
mRNA=t(as.matrix(BRCA.mRNA[,-1]))
colnames(mRNA)=BRCA.mRNA[,1]
mRNA1=data.imputation(mRNA,fun="microarray")
mRNA1=FSbyMAD(mRNA1, cut.type="topk",value=5000)
###Split the normal and tumor samples
index=which(as.numeric(substr(colnames(mRNA1),14,15))>9)
mRNA_normal=mRNA1[,index]
mRNA_tumor=mRNA1[,-index]
### Remove the duplicate samples
index1=which(as.numeric(substr(colnames(mRNA_tumor),14,15))>1)
mRNA_tumor=mRNA_tumor[,-index1]
##### Identify cancer subtypes
result=ExecuteCC(clusterNum=5,d=mRNA_tumor,maxK=5,clusterAlg="hc",distance="pearson",title="BRCA")
group=result$group
res=DiffExp.limma(Tumor_Data=mRNA_tumor,Normal_Data=mRNA_normal,group=group,topk=NULL,RNAseq=FALSE)CancerSubtypes包提供了一套癌症亚型分析工具和标准化的工作流分析方法,很大的方便我们对癌症亚型的探索和发现。
参考资料:
1.http://bioconductor.org/packages/release/bioc/vignettes/CancerSubtypes/inst/doc/CancerSubtypes-vignette.html
