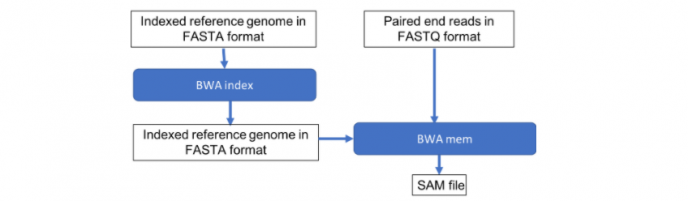
BWA – Burrows-Wheeler Alignment Tool ,是将测序序列快速、准确的比对到参考基因组上的开源软件。在二代或者三代测序种运用的相对比较多,尤其是其BWA-MEM 算法对illumina的数据效果更好。
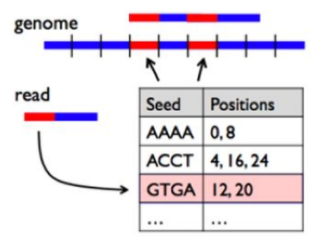
BWA通过创建参考序列的“索引”以加快查找速度,如左图: 显示了一个短的Read片段,其中有一个红色片段和一个蓝色片段,我们试图将其与包含许多蓝色和红色片段的基因组对齐。该索引记录了参考基因组中出现红色和蓝色片段(种子序列)给定模式的所有位置。当BWA遇到新的Read时,它将在索引中查找种子序列,并检索一组位置,这些位置是该Read的潜在比对位置,这样可以减少要检查匹配度的位置数,从而加快搜索速度。
目前BWA包含了三种算法:
- BWA-backtrack: designed for Illumina sequence reads up to 100bp (3-step)
- BWA-SW: designed for longer sequences ranging from 70bp to 1Mbp, long-read support and split alignment
- BWA-MEM: optimized for 70-100bp Illumina reads
BWA首先需要为参考基因组构建FM索引( index命令)。对齐算法由不同的子命令调用: aln / samse / sampe用于BWA-backtrack , bwasw用于BWA-SW, mem用于BWA-MEM算法。
常规BWA比对流程就大致可以分为两步:
- 建立索引
- 使用 BWA-backtrack 、 BWA-SW 、BWA-MEM(常用)进行比对
常用命令:
bwa index ref.fa
bwa mem ref.fa reads.fq > aln-se.sam
bwa mem ref.fa read1.fq read2.fq > aln-pe.sam
bwa aln ref.fa short_read.fq > aln_sa.sai
bwa samse ref.fa aln_sa.sai short_read.fq > aln-se.sam
bwa sampe ref.fa aln_sa1.sai aln_sa2.sai read1.fq read2.fq > aln-pe.sam
bwa bwasw ref.fa long_read.fq > aln.sam
1)建立索引
需要Ref序列文件,格式如下(标准的fasta文件格式,示例文件chr10.fa ):
>chr10 AC:CM000672.2 gi:568336… <-- '>' charachter followed by sequence name
NNNNNNNNNNNNNNNNNNNNN <-- sequence
…建立索引:bwa index chr10.fa生成如下文件
chr10.fa <-- Original sequence
chr10.fa.amb <-- Location of ambiguous (non-ATGC) nucleotides
chr10.fa.ann <-- Sequence names, lengths
chr10.fa.bwt <-- BWT suffix array
chr10.fa.pac <-- Binary encoded sequence
chr10.fa.sa <-- Suffix array index2)BWA alignment。此处我们默认使用 bwa mem 演示
Usage: bwa mem [options] <idxbase> <in1.fq> [in2.fq]
Algorithm options:
-t INT number of threads [1]
-k INT minimum seed length [19]
-w INT band width for banded alignment [100]
-d INT off-diagonal X-dropoff [100]
-r FLOAT look for internal seeds inside a seed longer than {-k} * FLOAT [1.5]
-y INT seed occurrence for the 3rd round seeding [20]
-c INT skip seeds with more than INT occurrences [500]
-D FLOAT drop chains shorter than FLOAT fraction of the longest overlapping chain [0.50]
-W INT discard a chain if seeded bases shorter than INT [0]
-m INT perform at most INT rounds of mate rescues for each read [50]
-S skip mate rescue
-P skip pairing; mate rescue performed unless -S also in use
...以双端测序结果为例:
bwa mem \
-t 2 \
-M \
-R "@RG\tID:reads\tSM:na12878\tPL:illumina" \
-o results/na12878.sam \
ref_data/chr10.fa \
raw_data/na12878_1.fq \
raw_data/na12878_2.fq-t 2 : BWA 使用两个线程
-M: 使用选项-M,一些 短的剪切体的匹配被标记为次要 alignment (不是主要 alignment , 会被大多数“旧”工具忽略 ) 。
-R "@RG\tID:reads\tSM:na12878\tPL:illumina" : 添加read group tag (RG), sample name (SM), 和platform (PL) 到比对结果的文件头上.
-o results/na12878.sam : 输出比对结果
生成的SAM文件(文件头):
@SQ SN:chr10 LN:133797422 <-- Reference sequence name (SN) and length (LN)
@RG ID:reads SM:na12878 <-- Read group (ID) and sample (SM) information that we provided
@PG ID:bwa PN:bwa VN:0.7.17… CL:bwa mem <-- Programs and arguments used in processing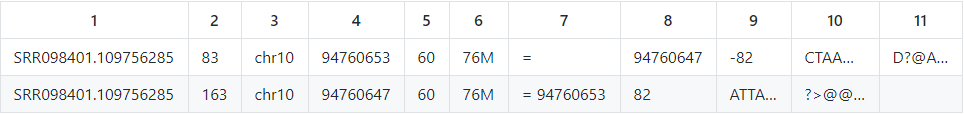
- Read ID
- Flag: 显示比对信息 例如: paired,aligned,等等。可以参考 decode flags.
- Reference sequence name
- 在参考序列中开始位置
- Mapping Quality
- CIGAR string: 比对总览信息如: match (M), insertion (I), deletion (D)
- RNEXT: 下一个片段比对上的参考序列的编号,没有另外的片段,这里是’*’,同一个片段,用’=’
- PNEXT: 下一个片段比对到参考序列上的第一个碱基位置
- TLEN: 模板长度,原始DNA或RNA片段的大小
- Read Sequence
- Read Quality
生成SAM文件后,可以用samtools等软件进行后续的下一步处理。
参考资料:
1.https://github.com/lh3/bwa
2.http://bio-bwa.sourceforge.net/bwa.shtml
3.https://rbatorsky.github.io/intro-to-ngs-bioinformatics/lessons/03_Alignment.html
