关于Blast算法和软件,我最早接触的时候是2013年左右,前后写过几次关于blast如何本地化的博文,后来由于站点从github迁移到云主机上(微信公众号上也写过《Blast本地化快速上手教程》),之前的写的内容大多丢失了,本文就旧瓶装新酒,再次介绍一下blast。

Basic Local Alignment Search Tool (Altschul et al., 1990 & 1997) ,blast算法采用了一种启发式算法,首先将输入的序列打断成子片段,称之为seed,然后将seed与预先索引好的序列进行比对,选择seed连续打分较高的位置采用动态规划算法进行延伸并且进行打分,当打分低于某一阈值延伸过程就会终止,最后产生了一系列高得分序列。最后使用E-value对其显著性进行评估,选出比对结果最好的序列。即Blast算法原理如下:

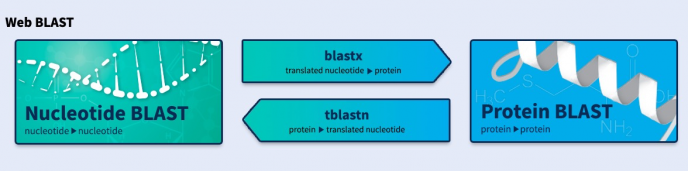
Blast+是目前比较常用的序列查找工具,包括核酸、蛋白序列的查找比对、同源性分析等。一般使用网页进行比对查询,如果有大量数据需要处理则需要安装独立程序进行分析,目前最新版本是BLAST+ 2.11.0,主要改进了对大数据的支持和新增了一些功能特性:更新历史。
独立运行程序下载地址:
https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/
Web版程序地址:
https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastp&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome
下面以linux下构建本地分析为例:
1)下载安装完成后,使用BLAST+自带的update_blastdb.pl脚本下载nr和nt等库文件,直接运行下列命令即可自动下载,或者fasta手动格式化数据库。
2)格式化数据库
示例:makeblastdb -in demo.fasta -dbtype prot -parse_seqids -out dbname
参数说明:
-in:待格式化的序列文件
-dbtype:数据库类型,prot或nucl
-out:数据库名
3)序列比对,以蛋白序列比对为例
blastp -query demo.fasta -out demo.blast -db dbname -outfmt 6 -evalue 1e-5 -num_descriptions 10 -num_threads 8
参数说明:
-query: 输入文件路径及文件名
-out:输出文件路径及文件名
-db:格式化了的数据库路径及数据库名
-outfmt:输出文件格式,总共有12种格式
-evalue:设置输出结果的e-value阈值
-num_threads:线程数
详细的参数建议多看看官方文档。
参考资料:
1.https://www.ncbi.nlm.nih.gov/books/NBK131777/
2.https://blast.ncbi.nlm.nih.gov/
3.https://www.ncbi.nlm.nih.gov/books/NBK51062/
