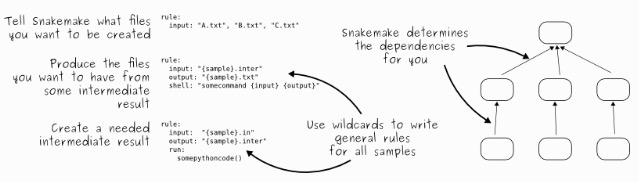
生信流程构建有很多方法,其中snakemake是诸多方法中比较小清新的一款流程搭建管理系统,和之前介绍的WDL《WDL(Workflow Description Language)搭建pipeline利器》有诸多相似之处,也有很多优势:
- 轻便,基于python构建,可以和python无缝衔接
- 支持任务投递系统,可以直接通过snakemake提交到任务系统
- 支持断点运行
- 支持多线程(这个功能基本的workflow管理工具都支持)
安装snakemake:
# 推荐用conda管理环境
conda create -n snakemake snakemake
# 创建好环境后,进入环境
conda activate snakemakesnakemake语法:
snakemake = statement | rule | include | workdir
rule = "rule" (identifier | "") ":" ruleparams
include = "include:" stringliteral
workdir = "workdir:" stringliteral
ni = NEWLINE INDENT
ruleparams = [ni input] [ni output] [ni params] [ni message] [ni threads] [ni (run | shell)] NEWLINE snakemake
input = "input" ":" parameter_list
output = "output" ":" parameter_list
params = "params" ":" parameter_list
log = "log" ":" parameter_list
benchmark = "benchmark" ":" statement
cache = "cache" ":" bool
message = "message" ":" stringliteral
threads = "threads" ":" integer
resources = "resources" ":" parameter_list
version = "version" ":" statement
run = "run" ":" ni statement
shell = "shell" ":" stringliteralsnakemake常用参数
--snakefile, -s 指定Snakefile,否则是当前目录下的Snakefile
--dryrun, -n 不真正执行,一般用来检查Snakefile是否有错
--printshellcmds, -p 输出要执行的shell命令
--reason, -r 输出每条rule执行的原因,默认FALSE
--cores, --jobs, -j 指定运行的核数,若不指定,则使用最大的核数
--force, -f 重新运行第一条rule或指定的rule
--forceall, -F 重新运行所有的rule,不管是否已经有输出结果
--forcerun, -R 重新执行Snakefile,当更新了rule时候使用此命令下面我们用一个简单的示例介绍其用法:
1)创建生信分析环境environment.yaml(推荐用conda创建和管理环境)
channels:
- conda-forge
- bioconda
dependencies:
- python =3.6
- jinja2 =2.10
- networkx =2.1
- matplotlib =2.2.3
- graphviz =2.38.0
- bcftools =1.9
- samtools =1.9
- bwa =0.7.17
- pysam =0.15.02)构建配置文件config.yaml(配置文件格式可以是JOSN、YAML)
samples:
A: data/samples/A.fastq
B: data/samples/B.fastq3)定义规则(workflow)
configfile: "config.yaml"
rule all:
input:
"plots/quals.svg"
rule bwa_map: # rule关键字,定义每个pipe
input: # 定义输入
"data/genome.fa",
lambda wildcards: config["samples"][wildcards.sample]
output: # 定义输出
temp("mapped_reads/{sample}.bam")
conda: # 选择环境
"environment.yaml"
params: # 参数设置
rg=r"@RG\tID:{sample}\tSM:{sample}"
log: # 日志
"logs/bwa_mem/{sample}.log"
threads: 8 # 线程数
shell: # shell命令,也可以用script来执行脚本,或者直接用run来执行python代码
"(bwa mem -R '{params.rg}' -t {threads} {input} | "
"samtools view -Sb - > {output}) 2> {log}"
rule samtools_sort:
input:
"mapped_reads/{sample}.bam"
output:
protected("sorted_reads/{sample}.bam")
conda:
"environment.yaml"
shell:
"samtools sort -T sorted_reads/{wildcards.sample} "
"-O bam {input} > {output}"
rule samtools_index:
input:
"sorted_reads/{sample}.bam"
output:
"sorted_reads/{sample}.bam.bai"
conda:
"environment.yaml"
shell:
"samtools index {input}"
rule bcftools_call:
input:
fa="data/genome.fa",
bam=expand("sorted_reads/{sample}.bam", sample=config["samples"]),
bai=expand("sorted_reads/{sample}.bam.bai", sample=config["samples"])
output:
"calls/all.vcf"
conda:
"environment.yaml"
shell:
"samtools mpileup -g -f {input.fa} {input.bam} | "
"bcftools call -mv - > {output}"
rule plot_quals:
input:
"calls/all.vcf"
output:
"plots/quals.svg"
conda:
"environment.yaml"
script:
"scripts/plot-quals.py"4)执行任务
snakemake --use-conda --snakefile snakefile参考资料:
1.https://snakemake.readthedocs.io/
