随着测序技术的发展,单细胞转录组测序(single-cell RNA-seq,scRNA-seq)技术得到了蓬勃的发展,可在单细胞水平揭示全基因组范围内所有基因的表达情况,有助于研究细胞间的表达异质性。目前已经有几十种不同的单细胞转录组测序技术相继被开发出来。

2017年的一篇 Molecular Cell 也详细比较比较了各个单细胞测序方法的异同:
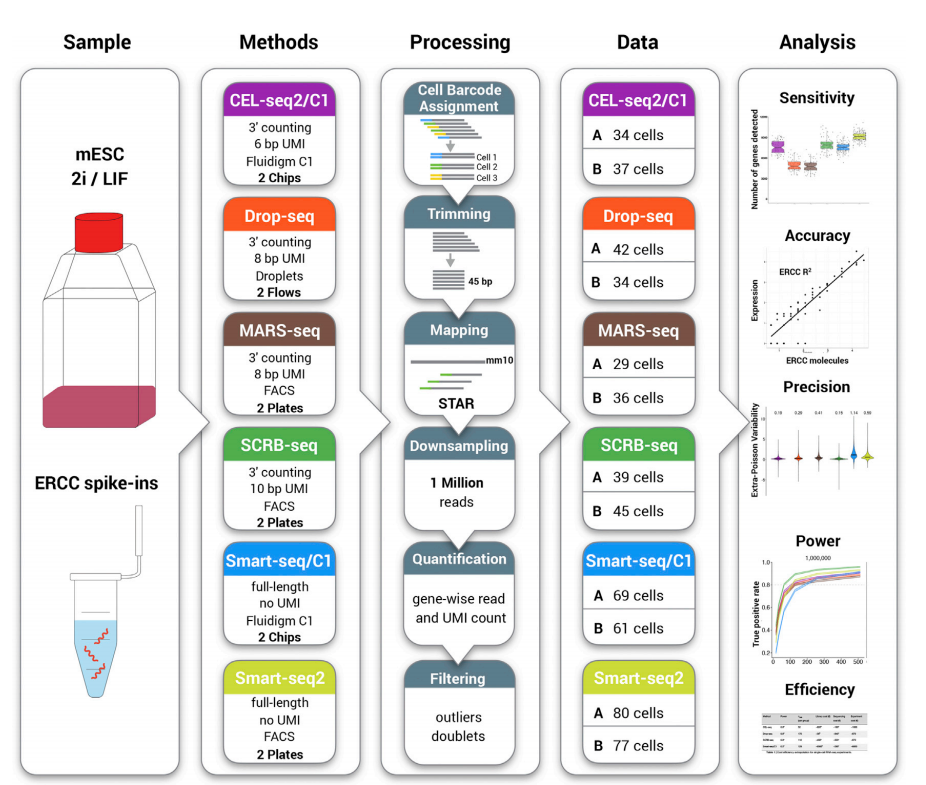

其中Smart-seq2作为测全长的转录本(full-length transcript sequencing)技术其中的一种,是由Picelli等人从Smart-seq中改良而来其对原始的Smart-Seq实验流程进行了多项改进优化,它不再需要纯化步骤,可大大提高产量。
Smart-seq2原理:通过设计oligo(dT)VN Primer作为逆转录引物,利用MMLVRT的模板转换活性,在cDNA的3’端添加一段接头序列,通过该接头序列进行反转录,生成cDNA第一条链。当逆转录酶到达mRNA5’末端时,会连续在末端添加几个胞嘧啶(C)残基。然后添加TSO(template-switching oligo)引物,退火后结合在第一条链的3’端与poly(C)突出杂交,合成第二条链。这样得到的cDNA经过PCR扩增,然后再纯化后用于测序。
Smart-seq2数据分析流程:
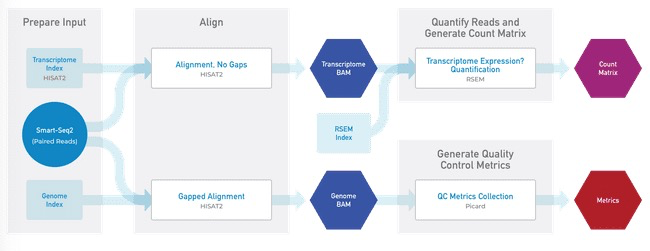
比对和组装及QC需要用到的软件:
| Pipeline Features | Description | Source |
|---|---|---|
| Assay Type | paired-end plate-based Smart-seq2 | |
| Overall workflow | Quality control module and transcriptome quantification module | Code available from Github |
| Workflow language | WDL | openWDL |
| Genomic reference sequence | GRCh38 human genome primary sequence | GENCODE |
| Gene Model | GENCODE v27 PRI GTF and Fasta files | GENCODE |
| Aligner | HISAT2 | Kim, et al.,2015; HISAT2 tool |
| QC | Metrics determined using Picard command line tools | Picard Tools |
| Estimation of gene expression | RSEM (rsem-calculate-expression) is used to estimate the gene expression profile. The input of RSEM is a bam file aligned by HISAT2. | Li and Dewey, 2011 |
| Data Input File Format | File format in which sequencing data is provided | FASTQ |
| Data Output File Format | File formats in which Smart-seq2 pipeline output is provided | BAM, Zarr version 2 |
常规scRNA-seq分析流程:
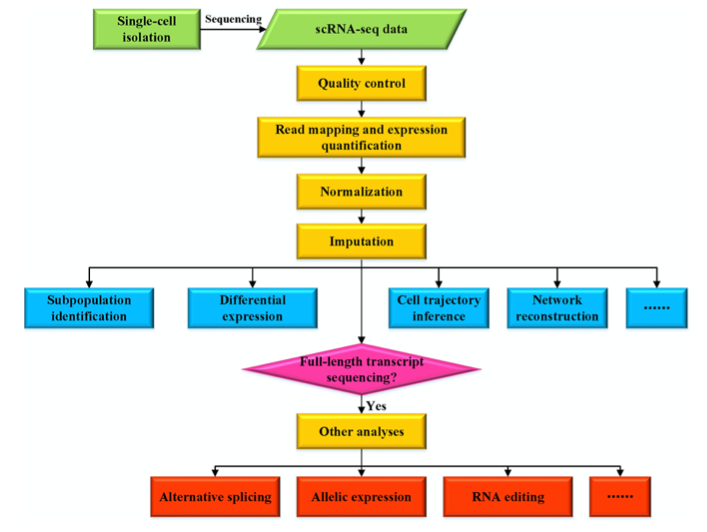
skylab提供了基于WDL语言的Workflow分析脚本,涵盖了下机原始数据的组装、以及质控部分,具体可以在github上招到其官方流程:
后续的分析培训教程可以参考之前的文章:scRNA-seq单细胞分析培训资源汇总,软件方法可以参考:单细胞资源大汇总。
参考资料:
1.https://data.humancellatlas.org/pipelines/smart-seq2-workflow
2.https://learn.gencore.bio.nyu.edu/single-cell-rnaseq/
3.Chen G, Ning B, Shi T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front Genet. 2019;10:317.
4. Ziegenhain C, Vieth B, Parekh S, et al. Comparative Analysis of Single-Cell RNA Sequencing Methods[J]. Molecular Cell, 2017, 65(4):631-643.e4.
