最近两年兴起了很多DIA分析的软件,尤其是Library free的方法,像PECAN、Prosit、DIA-NN等基于深度学习的方法,但是不得不说一个方法DIA-Umpire,利用了多肽在MS1下的打碎效率非100%的一个特点,即部分未碎片化的多肽和碎裂后的子离子也会进入MS2,然后基于RT计算一个相关性从而推导出可能的对应的母离子(多肽)和子离子的关系,从而构建一个虚拟的Library,后续的分析就和常规的DIA分析一样了,大名鼎鼎的Spectronaut的Library free就是基于此方法。

这里面提到了DIA数据的分析两种策略:
1)一种是试图将肽与单个谱图(以谱图为中心,Spectrum-Centric)匹配,DDA方式,直接用多肽序列和单个谱图进行Match;
2)另一种是试图检测数据文件中某个给定的肽(以肽为中心,Peptide-Centric)匹配,DIA方法,是基于Libray检测给定的多肽(我的理解是碎片肽段离子和多肽的一个匹配)。
DIA-Umpire主要原理:
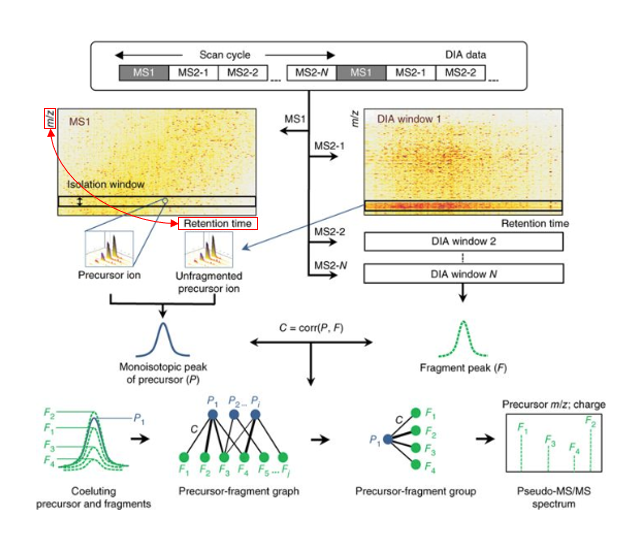
简单来说,就是我们分析DDA直接用fasta是因为,DDA的窄的窗口很大程度上的保留了MS1和MS2的对应关系,我们直接用Fasta序列和单个谱图进行Match就可以得到多肽和蛋白的信息,但是DIA分析中这种关系就不存在了;DIA-Umpire巧妙的利用多肽在MS1下的打碎效率非100%的一个特点 ,计算出MS1和那些未碎片化的多肽的一个相关性,从而间接找出MS1和MS2的对应关系,通过这种方式建立一个假库(Pseudo-Library),从而实现Library free的方式,不得不说这个方法是非常的巧妙的。
主要包含了以下几个模块:
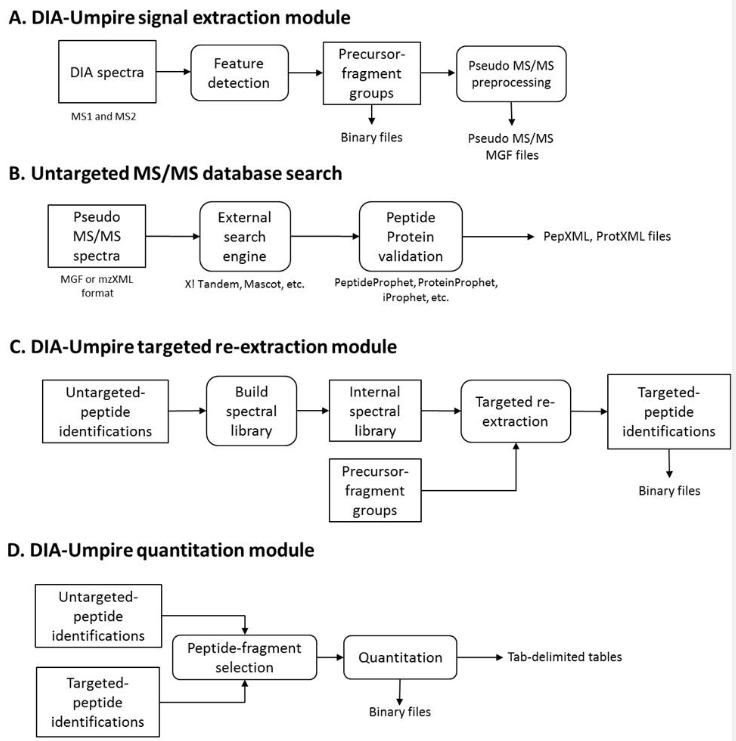
下面是它的一个简单的工作流程:
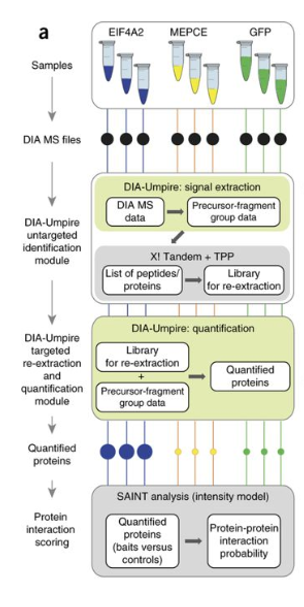
方法和思路都较为经典,学习Library free的很好的素材。
参考资料:
1.https://diaumpire.nesvilab.org/
2.https://github.com/Nesvilab/DIA-Umpire
3.https://sourceforge.net/projects/diaumpire/files/
4.https://github.com/cctsou/DIA-Umpire
5. Tsou, C., Avtonomov, D., Larsen, B. et al. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat Methods12, 258–264 (2015).
