最近看见一个新的蛋白组差异蛋白分析的R包,和大家分享。DEqMS是基于Limma开发,在Limma包上做了很多优化来适应蛋白组学数据分析。其中,Limma假设所有基因具有相同的先验方差而在蛋白质组学中,蛋白质丰度估计值的准确性受到label-free和 labeled方法 中量化的多肽/PSM的数量而异。作者认为多肽或PSM对蛋白质的定量更为准确,DEqMS包能够估计由不同数量的PSM/多肽定量的蛋白质的不同先验方差,具有更好的准确性,该R包可用来分析 label-free和 labeled 的蛋白质组数据。
下面我们用作者的一个简单示例给大家演示一下分析过程:
# 安装DEqMS包
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("DEqMS")作者示例中展示了一个基于 TMT10plex 的蛋白组学数据,用了三种不同miRNA类似物处理A431细胞(人表皮状癌细胞系)( Zhou Y. Et al Oncogene 2017 ),原始MS数据用 MS-GF+ 搜索,用Percolator后处理,并使用1% protein level FDR过滤得到的蛋白矩阵。
大家可以下载上面的蛋白数据,也可以用R下载:
# Pride ID:PXD004163
url <- "ftp://ftp.pride.ebi.ac.uk/pride/data/archive/2016/06/PXD004163/Yan_miR_Protein_table.flatprottable.txt"
download.file(url, destfile = "./miR_Proteintable.txt",method = "auto")下面我们用miR_Proteintable.txtTMT数据做后续的差异蛋白分析
# 载入library
library(DEqMS)
library(matrixStats)
# 读取数据
df.prot = read.table(
"miR_Proteintable.txt",
stringsAsFactors = FALSE,
header = TRUE,
quote = "",
comment.char = "",
sep = "\t"
)
# 蛋白水平FDR过滤<0.01
## 提取TMT蛋白定量信息
TMT_columns = seq(15, 33, 2)
dat = df.prot[df.prot$miR.FASP_q.value < 0.01, TMT_columns]
rownames(dat) = df.prot[df.prot$miR.FASP_q.value < 0.01, ]$Protein.accession
# 取log2,删除NA
dat.log = log2(dat)
dat.log = na.omit(dat.log)
# 绘制盒状图
boxplot(dat.log, las = 2, main = "TMT10plex data PXD004163")
接下来设计分组,这个采用limma的设置分组的方法:
cond = as.factor(
c(
"ctrl",
"miR191",
"miR372",
"miR519",
"ctrl",
"miR372",
"miR519",
"ctrl",
"miR191",
"miR372"
)
)
# 设计实验
design = model.matrix( ~ 0 + cond)
colnames(design) = gsub("cond", "", colnames(design))
# 定义分组比较
x <- c(
"miR372-ctrl",
"miR519-ctrl",
"miR191-ctrl",
"miR372-miR519",
"miR372-miR191",
"miR519-miR191"
)
contrast = makeContrasts(contrasts = x, levels = design)接下来这一步可能相对比较重要了这也是作者R包的思想或者精髓所在,作者先用limma计算了差异,然后作者统计了PSM的信息:
# 采用limma计算,并进行经验贝叶斯修正计算P
fit1 <- lmFit(dat.log, design)
fit2 <- contrasts.fit(fit1, contrasts = contrast)
fit3 <- eBayes(fit2)
# 添加PSM信息
count_columns = seq(16, 34, 2)
psm.count.table = data.frame(count = rowMins(as.matrix(df.prot[, count_columns])),
row.names = df.prot$Protein.accession)
fit3$count = psm.count.table[rownames(fit3$coefficients), "count"]
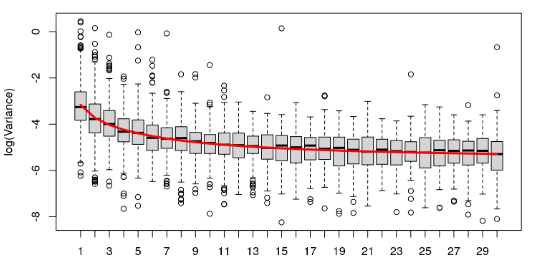
fit4 = spectraCounteBayes(fit3)作者用方差和PSM展示数据的真实分布,此处选择PSM<30:
# <= 30 PSMs.
VarianceBoxplot(fit4,
n = 30,
main = "TMT10plex dataset PXD004163",
xlab = "PSM count")
VarianceScatterplot(fit4, main = "TMT10plex dataset PXD004163")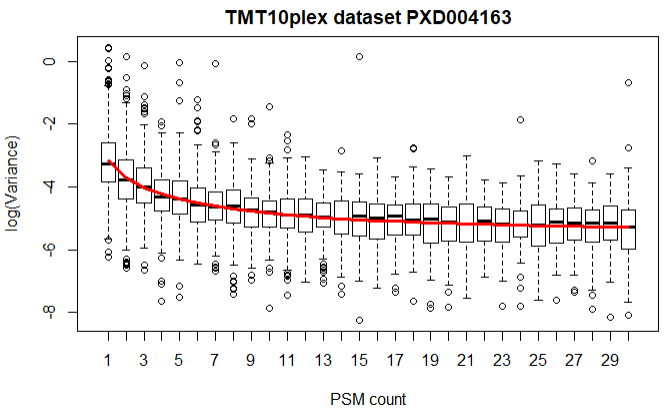

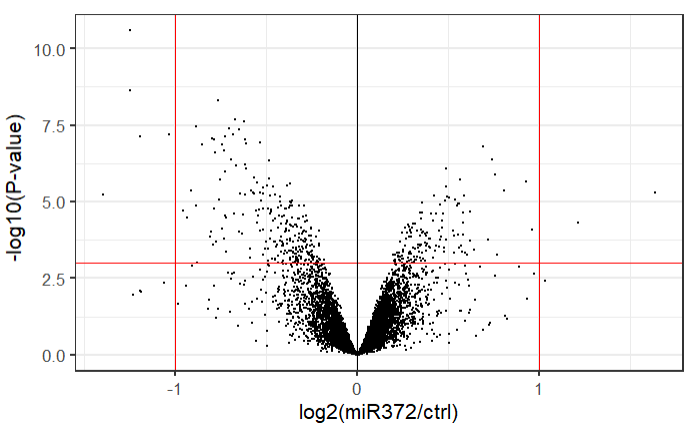
后续绘图和常规火山图类似,此处不在赘述:
library(ggrepel)
# ggplot2绘制火山图
DEqMS.results$log.sca.pval = -log10(DEqMS.results$sca.P.Value)
ggplot(DEqMS.results, aes(x = logFC, y = log.sca.pval)) +
geom_point(size = 0.5) +
theme_bw(base_size = 16) + # change theme
xlab(expression("log2(miR372/ctrl)")) + # x-axis label
ylab(expression(" -log10(P-value)")) + # y-axis label
geom_vline(xintercept = c(-1, 1), colour = "red") + # Add fold change cutoffs
geom_hline(yintercept = 3, colour = "red") + # Add significance cutoffs
geom_vline(xintercept = 0, colour = "black") + # Add 0 lines
scale_colour_gradient(low = "black", high = "black", guide = FALSE) +
geom_text_repel(data = subset(DEqMS.results, abs(logFC) > 1 &
log.sca.pval > 3),
aes(logFC, log.sca.pval , label = gene)) # add gene label
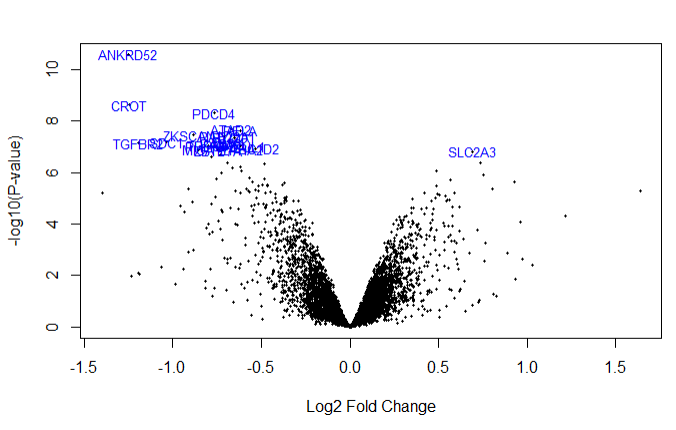
fit4$p.value = fit4$sca.p
# limma包自带volcanoplot函数绘制火山图
volcanoplot(
fit4,
coef = 1,
style = "p-value",
highlight = 20,
names = rownames(fit4$coefficients)
)
同时作者还同t-test、 ANOVA 和 Limma做了比较,大家可以查看参考资料1获取详细的信息。
参考资料:
1.http://www.bioconductor.org/packages/release/bioc/vignettes/DEqMS/inst/doc/DEqMS-package-vignette.html
