
X! Tandem是一个开源的蛋白质谱数据搜索引擎,支持DTA、PKL 、MGF 质谱产生的二进制文件。关于 X! Tandem 的详细文章发表在2004年左右,大家感兴趣可以在https://www.ncbi.nlm.nih.gov/pubmed/14976030处查到相关文章。
1)首先至文章下载 X! Tandem 可执行程序,linux版请至官网下载源码编译。
文件结构如下,官方程序包含一个实例,使用很简单主要在各项参数的调整上面,后续大家可以根据实例进行学习:

2)运行示例:
# windows 下执行,操作很简单
tandem.exe input.xml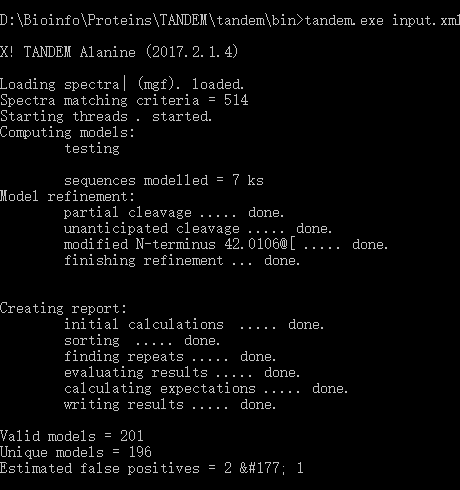
3)详细说明

input.xml文件,主要包含三块:default_input.xml是软件的各项参数内容;taxonomy.xml是指定我们的fasta.pro文件(稍后我们介绍fasta_pro软件的使用);另一个是我们需要指定我们的待解析的质谱数据文件test_spectra.mgf
<?xml version="1.0"?>
<bioml>
<note>
Each one of the parameters for x! tandem is entered as a labeled note node.
Any of the entries in the default_input.xml file can be over-ridden by
adding a corresponding entry to this file. This file represents a minimum
input file, with only entries for the default settings, the output file
and the input spectra file name.
See the taxonomy.xml file for a description of how FASTA sequence list
files are linked to a taxon name.
</note>
<note type="input" label="list path, default parameters">default_input.xml</note>
<note type="input" label="list path, taxonomy information">taxonomy.xml</note>
<note type="input" label="protein, taxon">yeast</note>
<note type="input" label="spectrum, path">test_spectra.mgf</note>
<note type="input" label="output, path">output.xml</note>
<note type="input" label="output, results">valid</note>
</bioml>default_input.xml 包含了各项的搜索配置,参数详细请读者仔细查看note字段的参数介绍,也可以至参考文章处查看文档,此处不再赘述每一个参数。
<?xml version="1.0"?>
<?xml-stylesheet type="text/xsl" href="tandem-input-style.xsl"?>
<bioml>
<note>list path parameters</note>
<note type="input" label="list path, default parameters">default_input.xml</note>
<note>This value is ignored when it is present in the default parameter
list path.</note>
<note type="input" label="list path, taxonomy information">taxonomy.xml</note>
<note>spectrum parameters</note>
<note type="input" label="spectrum, fragment monoisotopic mass error">0.4</note>
<note type="input" label="spectrum, parent monoisotopic mass error plus">100</note>
<note type="input" label="spectrum, parent monoisotopic mass error minus">100</note>
<note type="input" label="spectrum, parent monoisotopic mass isotope error">yes</note>
<note type="input" label="spectrum, fragment monoisotopic mass error units">Daltons</note>
<note>The value for this parameter may be 'Daltons' or 'ppm': all other values are ignored</note>
<note type="input" label="spectrum, parent monoisotopic mass error units">ppm</note>
<note>The value for this parameter may be 'Daltons' or 'ppm': all other values are ignored</note>
<note type="input" label="spectrum, fragment mass type">monoisotopic</note>
<note>values are monoisotopic|average </note>
<note>spectrum conditioning parameters</note>
<note type="input" label="spectrum, dynamic range">100.0</note>
<note>The peaks read in are normalized so that the most intense peak
is set to the dynamic range value. All peaks with values of less that
1, using this normalization, are not used. This normalization has the
overall effect of setting a threshold value for peak intensities.</note>
<note type="input" label="spectrum, total peaks">50</note>
<note>If this value is 0, it is ignored. If it is greater than zero (lets say 50),
then the number of peaks in the spectrum with be limited to the 50 most intense
peaks in the spectrum. X! tandem does not do any peak finding: it only
limits the peaks used by this parameter, and the dynamic range parameter.</note>
<note type="input" label="spectrum, maximum parent charge">4</note>
<note type="input" label="spectrum, use noise suppression">yes</note>
<note type="input" label="spectrum, minimum parent m+h">500.0</note>
<note type="input" label="spectrum, minimum fragment mz">150.0</note>
<note type="input" label="spectrum, minimum peaks">15</note>
<note type="input" label="spectrum, threads">1</note>
<note type="input" label="spectrum, sequence batch size">1000</note>
<note>residue modification parameters</note>
<note type="input" label="residue, modification mass">57.022@C</note>
<note>The format of this parameter is m@X, where m is the modfication
mass in Daltons and X is the appropriate residue to modify. Lists of
modifications are separated by commas. For example, to modify M and C
with the addition of 16.0 Daltons, the parameter line would be
+16.0@M,+16.0@C
Positive and negative values are allowed.
</note>
<note type="input" label="residue, potential modification mass"></note>
<note>The format of this parameter is the same as the format
for residue, modification mass (see above).</note>
<note type="input" label="residue, potential modification motif"></note>
<note>The format of this parameter is similar to residue, modification mass,
with the addition of a modified PROSITE notation sequence motif specification.
For example, a value of 80@[ST!]PX[KR] indicates a modification
of either S or T when followed by P, and residue and the a K or an R.
A value of 204@N!{P}[ST]{P} indicates a modification of N by 204, if it
is NOT followed by a P, then either an S or a T, NOT followed by a P.
Positive and negative values are allowed.
</note>
<note>protein parameters</note>
<note type="input" label="protein, taxon">other mammals</note>
<note>This value is interpreted using the information in taxonomy.xml.</note>
<note type="input" label="protein, cleavage site">[RK]|{P}</note>
<note>this setting corresponds to the enzyme trypsin. The first characters
in brackets represent residues N-terminal to the bond - the '|' pipe -
and the second set of characters represent residues C-terminal to the
bond. The characters must be in square brackets (denoting that only
these residues are allowed for a cleavage) or french brackets (denoting
that these residues cannot be in that position). Use UPPERCASE characters.
To denote cleavage at any residue, use [X]|[X] and reset the
scoring, maximum missed cleavage site parameter (see below) to something like 50.
</note>
<note type="input" label="protein, modified residue mass file"></note>
<note type="input" label="protein, cleavage C-terminal mass change">+17.002735</note>
<note type="input" label="protein, cleavage N-terminal mass change">+1.007825</note>
<note type="input" label="protein, N-terminal residue modification mass">0.0</note>
<note type="input" label="protein, C-terminal residue modification mass">0.0</note>
<note type="input" label="protein, homolog management">no</note>
<note>if yes, an upper limit is set on the number of homologues kept for a particular spectrum</note>
<note>model refinement parameters</note>
<note type="input" label="refine">yes</note>
<note type="input" label="refine, modification mass"></note>
<note type="input" label="refine, sequence path"></note>
<note type="input" label="refine, tic percent">20</note>
<note type="input" label="refine, spectrum synthesis">yes</note>
<note type="input" label="refine, maximum valid expectation value">0.1</note>
<note type="input" label="refine, potential N-terminus modifications">+42.010565@[</note>
<note type="input" label="refine, potential C-terminus modifications"></note>
<note type="input" label="refine, unanticipated cleavage">yes</note>
<note type="input" label="refine, potential modification mass"></note>
<note type="input" label="refine, point mutations">no</note>
<note type="input" label="refine, use potential modifications for full refinement">no</note>
<note type="input" label="refine, point mutations">no</note>
<note type="input" label="refine, potential modification motif"></note>
<note>The format of this parameter is similar to residue, modification mass,
with the addition of a modified PROSITE notation sequence motif specification.
For example, a value of 80@[ST!]PX[KR] indicates a modification
of either S or T when followed by P, and residue and the a K or an R.
A value of 204@N!{P}[ST]{P} indicates a modification of N by 204, if it
is NOT followed by a P, then either an S or a T, NOT followed by a P.
Positive and negative values are allowed.
</note>
<note>scoring parameters</note>
<note type="input" label="scoring, minimum ion count">4</note>
<note type="input" label="scoring, maximum missed cleavage sites">1</note>
<note type="input" label="scoring, x ions">no</note>
<note type="input" label="scoring, y ions">yes</note>
<note type="input" label="scoring, z ions">no</note>
<note type="input" label="scoring, a ions">no</note>
<note type="input" label="scoring, b ions">yes</note>
<note type="input" label="scoring, c ions">no</note>
<note type="input" label="scoring, cyclic permutation">no</note>
<note>if yes, cyclic peptide sequence permutation is used to pad the scoring histograms</note>
<note type="input" label="scoring, include reverse">no</note>
<note>if yes, then reversed sequences are searched at the same time as forward sequences</note>
<note type="input" label="scoring, cyclic permutation">no</note>
<note type="input" label="scoring, include reverse">no</note>
<note>output parameters</note>
<note type="input" label="output, log path"></note>
<note type="input" label="output, message">testing 1 2 3</note>
<note type="input" label="output, one sequence copy">no</note>
<note type="input" label="output, sequence path"></note>
<note type="input" label="output, path">output.xml</note>
<note type="input" label="output, sort results by">protein</note>
<note>values = protein|spectrum (spectrum is the default)</note>
<note type="input" label="output, path hashing">yes</note>
<note>values = yes|no</note>
<note type="input" label="output, xsl path">tandem-style.xsl</note>
<note type="input" label="output, parameters">yes</note>
<note>values = yes|no</note>
<note type="input" label="output, performance">yes</note>
<note>values = yes|no</note>
<note type="input" label="output, spectra">yes</note>
<note>values = yes|no</note>
<note type="input" label="output, histograms">yes</note>
<note>values = yes|no</note>
<note type="input" label="output, proteins">yes</note>
<note>values = yes|no</note>
<note type="input" label="output, sequences">yes</note>
<note>values = yes|no</note>
<note type="input" label="output, one sequence copy">no</note>
<note>values = yes|no, set to yes to produce only one copy of each protein sequence in the output xml</note>
<note type="input" label="output, results">valid</note>
<note>values = all|valid|stochastic</note>
<note type="input" label="output, maximum valid expectation value">0.1</note>
<note>value is used in the valid|stochastic setting of output, results</note>
<note type="input" label="output, histogram column width">30</note>
<note>values any integer greater than 0. Setting this to '1' makes cutting and pasting histograms
into spread sheet programs easier.</note>
<note type="description">ADDITIONAL EXPLANATIONS</note>
<note type="description">Each one of the parameters for X! tandem is entered as a labeled note
node. In the current version of X!, keep those note nodes
on a single line.
</note>
<note type="description">The presence of the type 'input' is necessary if a note is to be considered
an input parameter.
</note>
<note type="description">Any of the parameters that are paths to files may require alteration for a
particular installation. Full path names usually cause the least trouble,
but there is no reason not to use relative path names, if that is the
most convenient.
</note>
<note type="description">Any parameter values set in the 'list path, default parameters' file are
reset by entries in the normal input file, if they are present. Otherwise,
the default set is used.
</note>
<note type="description">The 'list path, taxonomy information' file must exist.
</note>
<note type="description">The directory containing the 'output, path' file must exist: it will not be created.
</note>
<note type="description">The 'output, xsl path' is optional: it is only of use if a good XSLT style sheet exists.
</note>
</bioml>
fasta.pro文件转换,此处我们根据官方提供的fasta_pro.exe可以转换
Usage: "fasta_pro filename [-i|-u|-n -d]"
-i force IPI file number output
-u force Unigene style description line processing
-d description line: enclose in quotes if there are spaces in the description注意:你也可以直接用fasta文件进行肽段的匹配搜索,官方推荐转化成优化后的pro文件,可能是更方便程序的读取。
4)文件类型介绍:
MGF 文件形如:
BEGIN IONS
PEPMASS=820.998855732003
CHARGE=1+
TITLE=Elution from: 0.14 to 0.14 period: 0 experiment: 2 cycles: 1
200.9942 2.3857
354.9856 2.3857
370.9314 5.1571
388.9714 9.6857
390.9608 2.7429
END IONS
BEGIN IONS
PEPMASS=691.910270874147
CHARGE=2+
TITLE=Elution from: 0.03 to 0.03 period: 0 experiment: 1 cycles: 1
264.8982 30.0286
264.9944 8.9429
435.8989 3.2857
442.9097 4.2571
478.9086 3.6571
END IONS解释:每个光谱都包含在一组BEGIN IONS和END IONS标记中。PEPMASS=后面的值是母体离子的质荷比。CHARGE之后的值是母离子电荷。TITLE后的是由空间分隔的子离子质量和强度对。
总的来说,现在有很多的软件可以进行肽段搜索,X! Tandem作为一款经典的肽段搜索软件也是值得大家探索和学习的。
软件下载:
参考文章:
1.https://thegpm.org/TANDEM/api/index.html
2.https://thegpm.org/TANDEM/tandem_install_faq.html
