MSstatsTMT的主要应用方向:
1.分析TMT标记的蛋白质组学数据
2.也支持代谢标记或iTRAQ标记。LC-MS, SRM, DIA(SWATH)与label-free等方法的蛋白质组学数据的分析。
总体来说软件还是很人性化的,支持多种软件的结果,作者的文章尚未发表,R包已经再github频繁更新了,预计近期可能online。下面简单介绍该包的功能及用法。
主要包含以下功能:
1.不同数据格式转换成 MSstatsTMT 需要的数据格式: PDtoMSstatsTMTFormat(Proteome Discoverer结果), MaxQtoMSstatsTMTFormat(maxquant结果), SpectroMinetoMSstatsTMTFormat( SpectroMine 格式结果) 和 OpenMStoMSstatsTMTFormat(OpenMS结果格式)
2.定量分析: proteinSummarization (蛋白定量信息)、 groupComparisonTMT (组间比较)
# 安装MSstatsTMT包
BiocManager::install("MSstatsTMT")下面我们用一个示例来看 MSstatsTMT 分析过程:
library(MSstatsTMT)
# 内置数据集,此处我们用Proteome discover的数据做演示
# data(raw.pd)
# 注释数据
# data(annotation.pd)
# Proteome discover数据转换成MSstatsTMT数据对象
input.pd <- PDtoMSstatsTMTFormat(raw.pd, annotation.pd)
# 移除缺失值
input.pd.no.miss <- PDtoMSstatsTMTFormat(raw.pd, annotation.pd,
rmPSM_withMissing_withinRun = TRUE)
# 用 MSstats方法计算蛋白的定量结果
quant.msstats <- proteinSummarization(input.pd,
method="msstats",
global_norm=TRUE,
reference_norm=TRUE,
remove_norm_channel = TRUE,
remove_empty_channel = TRUE)
# 用Median方法来计算蛋白定量结果
quant.median <- proteinSummarization(input.pd.no.miss,
method="Median",
global_norm=TRUE,
reference_norm=TRUE,
remove_norm_channel = TRUE,
remove_empty_channel = TRUE)
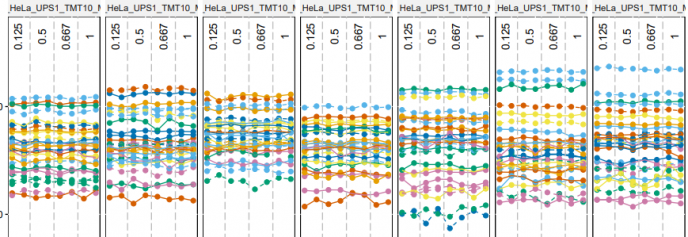
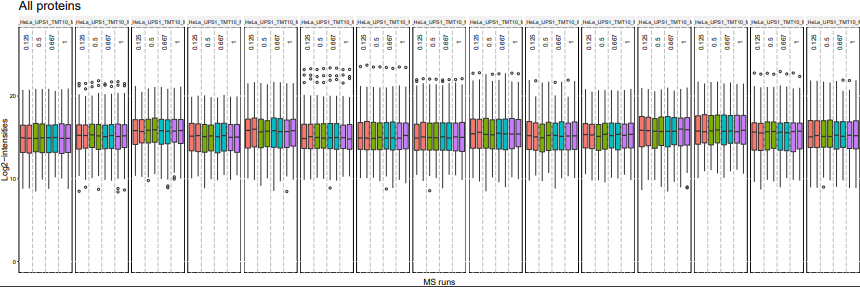
# QC 数据
dataProcessPlotsTMT(data.peptide = input.pd,
data.summarization = quant.msstats,
type = 'QCPlot',
width = 21,
height = 7)
# 默认比较所有分组
test.pairwise <- groupComparisonTMT(quant.msstats)
# 查看分组情况
levels(quant.msstats$Condition)
#> [1] "0.125" "0.5" "0.667" "1"

# 指定比较分组
# 只比较0.125 和 1
comparison<-matrix(c(-1,0,0,1),nrow=1)
row.names(comparison)<-"1-0.125"
# 设置列名
colnames(comparison)<- c("0.125", "0.5", "0.667", "1")
# 比较
test.contrast <- groupComparisonTMT(data = quant.msstats, contrast.matrix = comparison)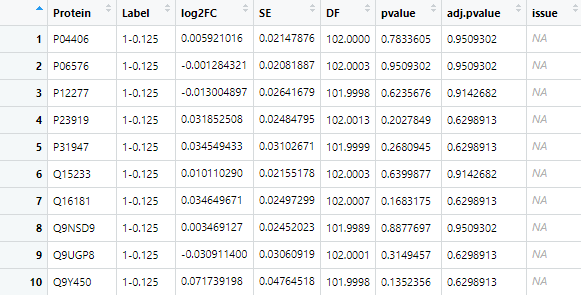
参考资料:
1.https://bioconductor.org/packages/release/bioc/vignettes/MSstatsTMT/inst/doc/MSstatsTMT.html
2.http://msstats.org/msstatstmt/
