
目前测序技术主要有单端测序(Single-read,目前已经逐步被双端测序取代)和双端测序(Paired-end),主流测序方法都是双端测序为主,其优势可以参见illumina官网。
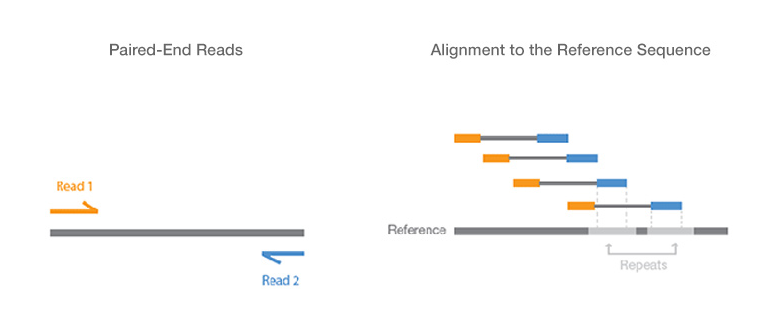
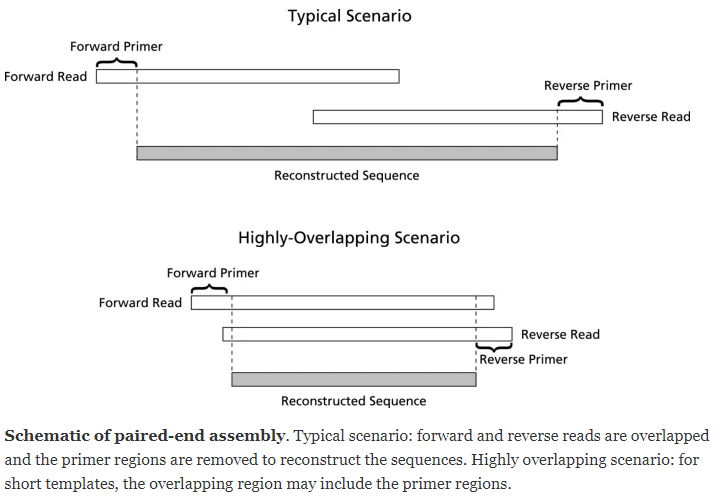
对于双端测序的原始数据结果一般会保存在在不同的 fastq 文件中,就是我们常见的*._R1.fq.gz,*._R2.fq.gz等形式。双端测序中每一个单独的 Read 其长度都超过整个待测基因的一半,根据两个 Reads 重合的部分对该序列进行拼接(PANDAseq拼接原理如下)组合成一个完整的fastq 文件:
1)安装PANDAseq
下载:https://github.com/neufeld/pandaseq
系统依赖(以Ubuntu为例):
sudo apt-get install build-essential libtool automake zlib1g-dev libbz2-dev pkg-config
# 编译安装
./autogen.sh && ./configure && make && sudo make install或者通过conda安装
conda install -c bioconda pandaseq2)软件使用
pandaseq -f forward.fastq -r reverse.fastq主要参数解释
-f 输入正向的 fastq 文件
-r 输入反向的 fastq 文件
-F 输出 fastq 文件
-T 线
参考资料:
1.https://github.com/neufeld/pandaseq
2.Masella, A.P., Bartram, A.K., Truszkowski, J.M. et al. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics 13, 31 (2012). https://doi.org/10.1186/1471-2105-13-31
3.https://www.illumina.com/science/technology/next-generation-sequencing/plan-experiments/paired-end-vs-single-read.html

Jaxonhe
你好,请问这个软件应该怎么在mac上面下载呢,我一直下载不好。如果可以的话,我想有偿请教一下这个问题。希望您通过邮件联系我
陈浩
你下载不好是指什么?编译报错?