
转录组分析工具可谓是数不胜数,今天介绍的一个R包-RNASeqR,可以一键解决转录组分析,下面简单介绍其部署及使用。
1)环境需求:
- R >= 3.5.0
- 需要安装 HISAT2、STAR 、StringTie和Gffcompare 并且加入系统的环境变量
- Python: Python2或者Python3
2)安装RNASeqR
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("RNASeqR")
# 安装示例数据
BiocManager::install("RNASeqRData")3)准备输入数据文件
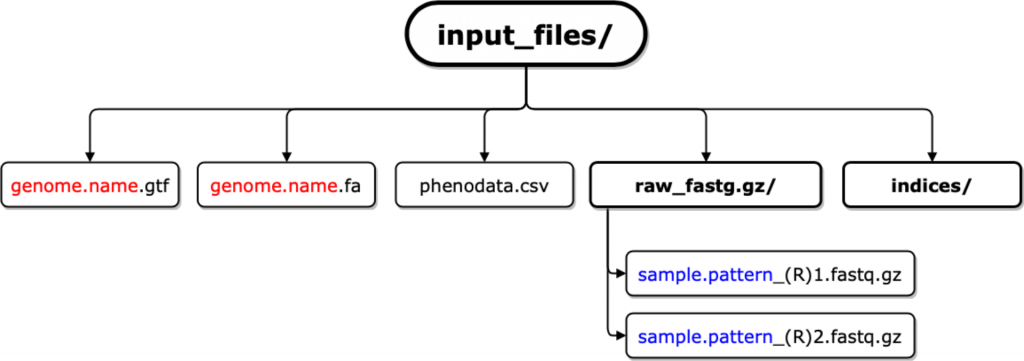
4)示例
library(RNASeqR)
library(RNASeqRData)
input_files.path <- system.file("extdata/", package = "RNASeqRData")
rnaseq_result.path <- "/tmp/RNASeqR/"
dir.create(rnaseq_result.path, recursive = TRUE)对于单端测序数据( “SE” ,single-end):
exp <- RNASeqRParam(path.prefix = rnaseq_result.path,
input.path.prefix = input_files.path,
genome.name = "Saccharomyces_cerevisiae_XV_Ensembl",
sample.pattern = "SRR[0-9]*_XV",
independent.variable = "state",
case.group = "60mins_ID20_amphotericin_B",
control.group = "60mins_ID20_control",
fastq.gz.type = "SE")对于双端测序数据( PE”,paired-end):
exp <- RNASeqRParam(path.prefix = rnaseq_result.path,
input.path.prefix = input_files.path,
genome.name = "Saccharomyces_cerevisiae_XV_Ensembl",
sample.pattern = "SRR[0-9]*_XV",
independent.variable = "state",
case.group = "60mins_ID20_amphotericin_B",
control.group = "60mins_ID20_control",
fastq.gz.type = "PE")序列比对
# 使用Hisat2进行比对
RNASeqReadProcess_CMD(exp, Hisat2.Index.run=TRUE,
Hisat2.Alignment.run = TRUE)
# 使用STAR进行
RNASeqReadProcess_CMD(exp, STAR.Alignment.run=TRUE,
Hisat2.Index.run=FALSE,
Hisat2.Alignment.run = FALSE)基因水平的差异分析
# 一键式
RNASeqDifferentialAnalysis_CMD(exp)基本可以做的常规的分析及图都有了,总体来说是比较便捷的。
参考资料:
1.https://www.bioconductor.org/packages/release/bioc/vignettes/RNASeqR/inst/doc/RNASeqR.html
