进化树在生物学中运用广泛,通常是用来表示物种之间的进化关系。生物分类学家往往根据各类生物间的亲缘关系的远近,把各类生物展示在有分枝的树状的图表上,简明地表示生物的进化历程和亲缘关系。其中使用比较广泛的一个工具是来自Segata实验室的GraPhlAn。
GraPhlAn是一种用于生成分类和系统发育树的综合可视化工具,其使用python语言编写而成,主要有graphlan.py和graphlan_annotate.py脚本。
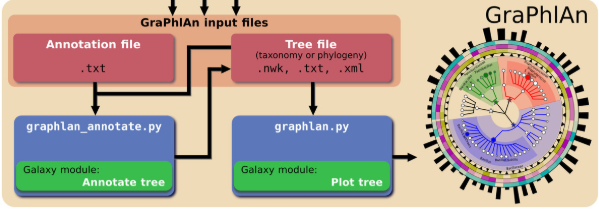
GraPhlAn结构如下图:
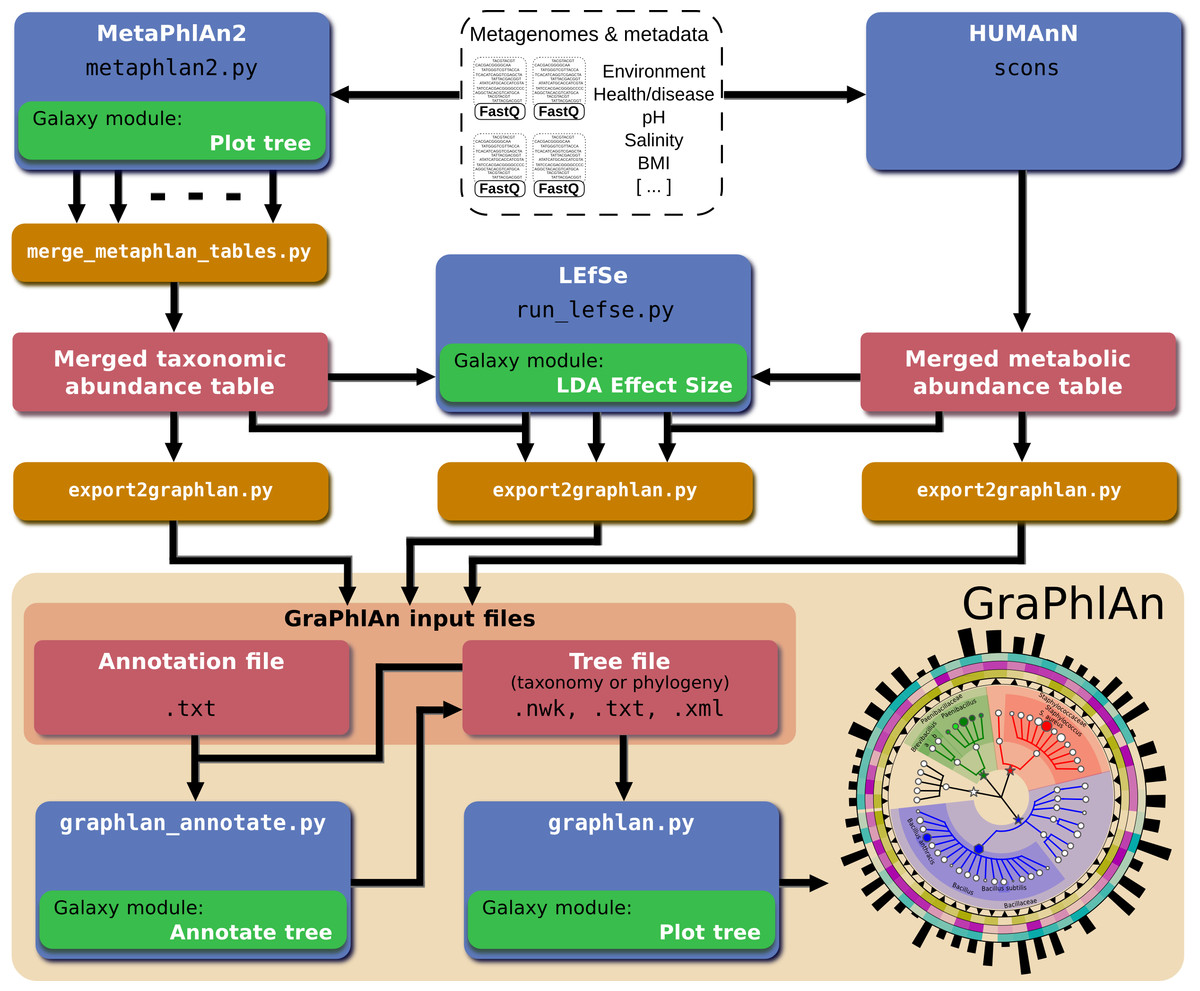
1)GraPhlAn安装:
- 通过 Bioconda:conda install -c bioconda graphlan
- 直接安装:pip install graphlan
- 从github安装(推荐,最新版的支持python3):
>git clone https://github.com/biobakery/graphlan.git
>pip install . - docker执行:docker run -it biobakery/graphlan bash
2)绘图
作者文章给出的示例也非常精美:
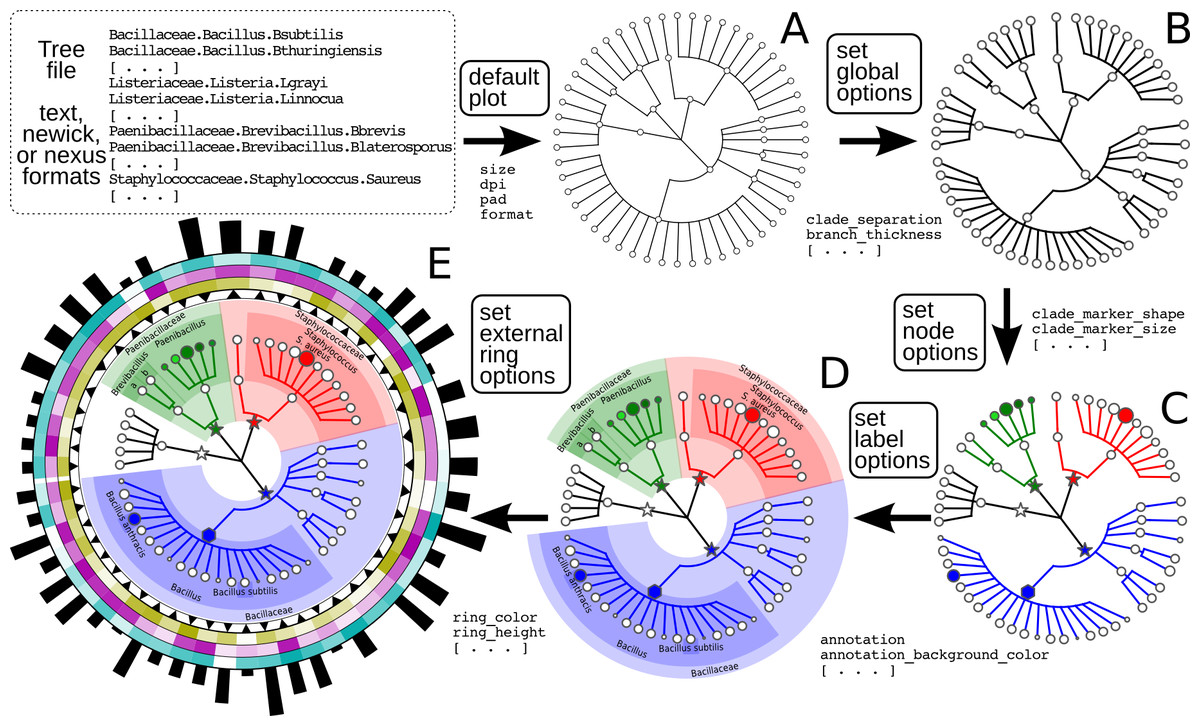
软件运行需要两个文件一个是构建进化树的文件一个是注释文件。
一般我们需要两步:
Step 1:合并树和注释文件-graphlan_annotate.py
usage: graphlan_annotate.py [-h] [--annot annotation_file] [-v]
input_tree [output_tree]
positional arguments:
input_tree 支持Newick, Nexus, PhyloXML或者文本文件
output_tree 输出含有注释的树文件,如果不指定将会覆盖源文件
optional arguments:
-h, --help 帮助
--annot annotation_file
注释文件
-v, --version 版本Step 2:绘制进化树-graphlan.py
usage: graphlan.py [-h] [--format ['output_image_format']]
[--warnings WARNINGS] [--positions POSITIONS]
[--dpi image_dpi] [--size image_size] [--pad pad_in]
[--external_legends] [--avoid_reordering] [-v]
input_tree output_image
positional arguments:
input_tree 输入文件,PhyloXML格式
output_image 输出图像名称,可以使用--format调整文件
格式: png, pdf, ps, eps, svg
optional arguments:
-h, --help 帮助
--format ['output_image_format']
指定图像格式
--warnings WARNINGS 警告
--positions POSITIONS
设置点的绝对位置是否应该显示到标准输出,r或theta
--dpi image_dpi 设置dpi
--size image_size 图片大小 (inche, 默认是 7.0)
--pad pad_in 图像的画布设置
--external_legends 图例设置(默认)
--avoid_reordering 指定树是否会重构
-v, --version 版本示例:利用作者提供的HMP_tree数据集
graphlan_annotate.py hmptree.xml hmptree.annot.xml --annot annot.txt
graphlan.py hmptree.annot.xml hmptree.png --dpi 150 --size 14 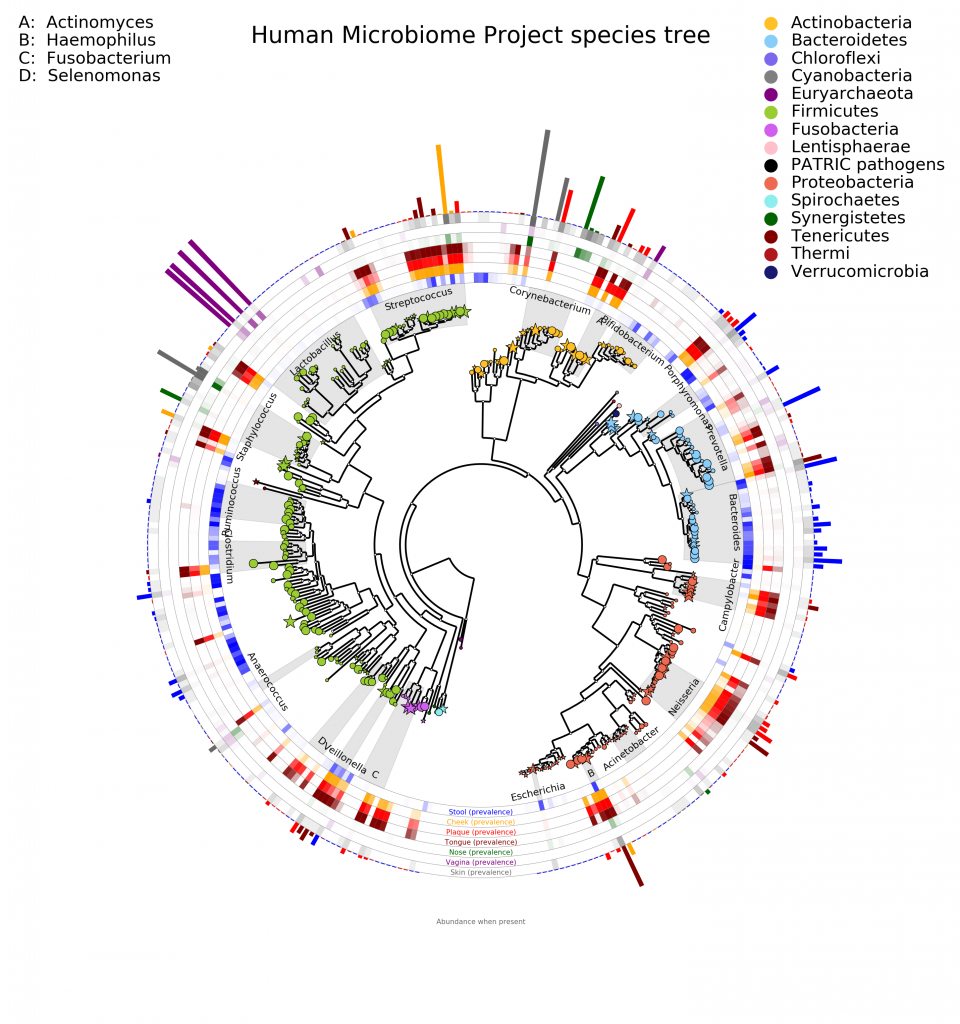
更多的参数介绍请查看参考资料。
参考资料:
1. https://github.com/biobakery/graphlan
2. https://github.com/biobakery/biobakery/wiki/graphlan
3. https://segatalab.github.io/tools/graphlan/
