
human_genomics_pipeline是一个基于bwa、gatk4的基因组Snakemake分析流程,可以用来处理双端测序数据。整个流程是从fastq处理到最后的vcf,可以利用GPU加速(NVIDIA GPU)。在配置文件中进行简单设置,指定特定物种的参考基因组和变异数据库来完成基因组的数据分析。
1)单个样本处理流程:
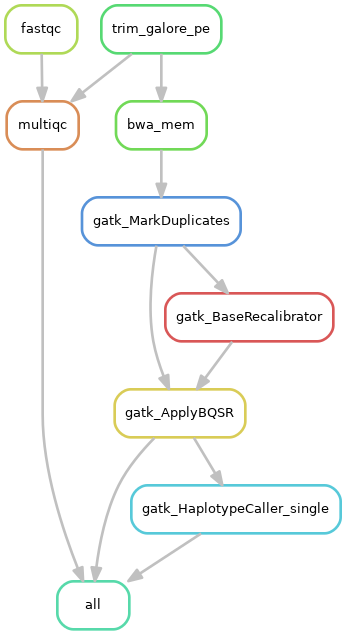
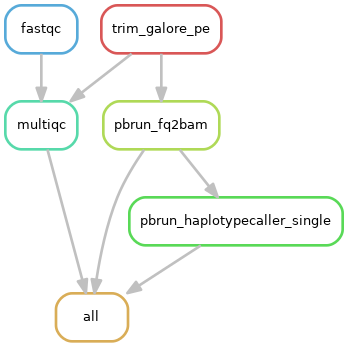
2)队列数据处理流程:

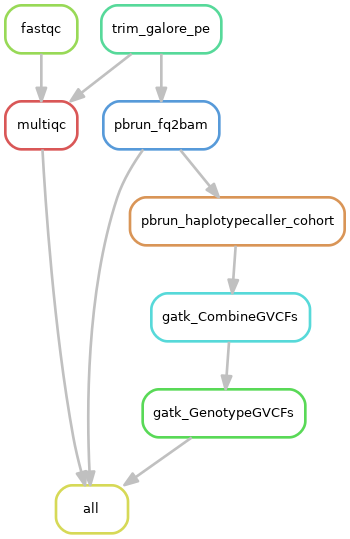
3)系统需求:
- 硬件: NVIDIA GPUs
- 硬件相关软件: NVIDIA CLARA PARABRICKS and dependencies , Git 2.7.4, Mamba 0.4.4 和 Conda 4.8.2, gsutil 4.52, gunzip 1.6
- 操作系统: Ubuntu 16.04
4)数据示例:
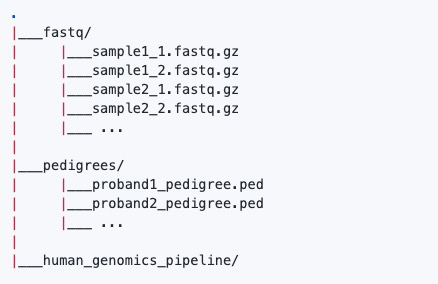
注意:输入双端测序fastq文件需要使用_1和_2标识,其他注意事项可以参考:访问
5)GATK resource bundle下载
参考:https://github.com/bahlolab/bioinfotools/blob/master/GATK/resource_bundle.md
6)配置文件(编辑config.yaml文件)
##############################
###### Overall workflow ######
##############################
# Type of input data (either 'Single' or 'Cohort')
DATA: ""
# Should the pipeline be GPU accelerated where possible? (either 'Yes' or 'No')
GPU_ACCELERATED: ""
# File path to the reference genome (.fasta)
REFGENOME: ""
# File path to dbSNP database
dbSNP: ""
# Temporary file directory
TEMPDIR: ""
# Whole exome sequence settings (leave blank if analysing other data such as whole genome sequence data)
WES:
# File path to the exome capture regions over which to operate (prefix with the '-L' flag)
INTERVALS: ""
# Padding (in bp) to add to each region (prefix with the '-ip' flag)
PADDING: ""
##############################
##### Pipeline resources #####
##############################
# Number of threads to use per rule/sample for multithreaded rules, multithreading will significantly speed up these rules (diminishing speed gains beyond 8 threads)
THREADS:
# Maximum memory usage per rule/sample (eg. '40g' for 40 gigabytes, this should suffice for exomes)
MAXMEMORY: ""
# Maximum number of GPU's to be used per rule/sample for gpu-accelerated runs (eg `1` for 1 GPU)
GPU:
##############################
########## Trimming ##########
##############################
# Whether or not to trim the raw fastq reads (either 'Yes' or 'No')
TRIM: ""
# If trimming, choose the adapter sequence to be trimmed (eg. `--illumina`, `--nextera` or `--small_rna`)
TRIMMING:
ADAPTERS: ""
##############################
##### Base recalibration #####
##############################
# Resources to used for base recalibration (prefix each resource with the '--known-sites' flag if not gpu accelerated and the '--knownSites' if gpu accelerated)
RECALIBRATION:
RESOURCES: ""HPC配置(cluster.json文件,可选):
{
"__default__" :
{
"account" : "",
"partition" : ""
}
}如果是使用是使用slurm,可以参考:如何在snakemake设置slurm
7)运行流程
当配置好配置文件就可以在bash下运行流程
bash dryrun.sh或者bash run.sh
# dryrun.sh内容
snakemake \
--dryrun \
--cores 32 \
--resources mem_mb=150000 \
--resources gpu=2 \
--use-conda \
--conda-frontend mamba \
--configfile ../config/config.yaml
# run.sh内容
snakemake \
--cores 32 \
--resources mem_mb=150000 \
--resources gpu=2 \
--use-conda \
--configfile ../config/config.yaml整个流程就介绍到此了,本流程的亮点是GPU的运用值得学习和借鉴。
参考资料:
1.https://github.com/ESR-NZ/human_genomics_pipeline
