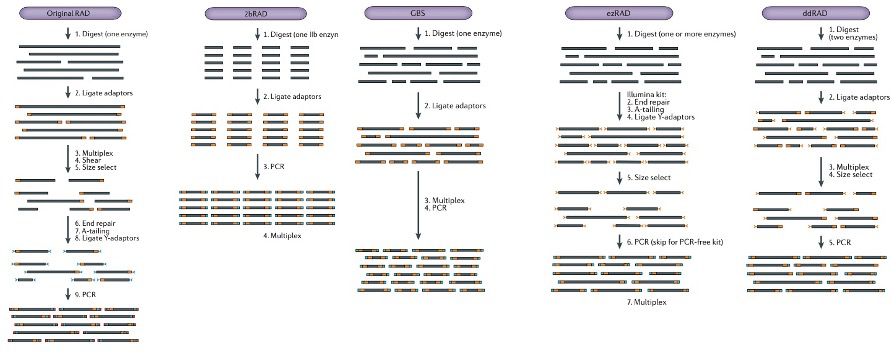
简化基因组测序(Reduced-Representation Genome Sequencing,RRGS)是指利用限制性内切酶打断基因组DNA,对特定片段进行高通量测序获得海量遗传多态性标签序列来充分代表目标物种全基因组信息的测序策略。此方法实验步骤简单,成本低,而且可以不依赖参考基因组,就能获得全基因组范围内的遗传多态性标签,因而广泛应用于生态学,进化学和基因组学等领域,目前常见如下几种技术:
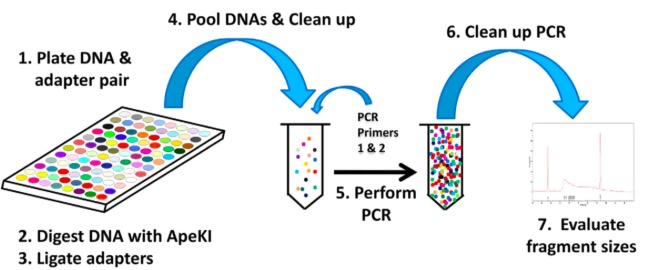
GBS(Genotyping-by-Sequencing)是简化基因组测序方法的一种。主要步骤如下:
- 将不同样本和含不同barcode接头成对放在平板里
- 使用ApeKI限制性内切酶进行酶解
- 使用T4连接酶,将接头连接到片段两端因酶切产生的粘末端
- 将含不同barcode的样本混池,随后过片段长度筛选柱,过滤尚未反应的接头
- 加入PCR引物,进行PCR扩增
GBS-SNP-CROP(GBS SNP Calling Reference Optional Pipeline)即:基于简化基因组测序的一个分析SNP的流程(其中参考序列是可选择的,即可以不用参考序列,流程会建立一个Mock Reference)该流程可以最大程度地利用GBS数据并在没有参考Reference的情况下执行SNP基因分型分析。
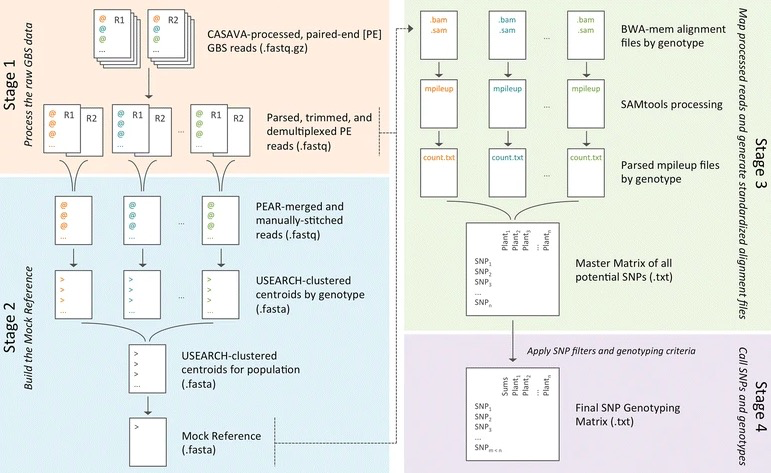
GBS-SNP-CROP用7个perl脚本建立了整个分析流程。其中用到很多关联软件,如:SAMtools、BWA等软件。GBS-SNP-CROP专为双端(PE)测序设计,很大限度地提高了数据使用率。
流程部署环境:
- Java 7 or higher – 最新版 GBS-SNP-CROP (v.4.1) 在java 8 (update 221)上正常使用
- Trimmomatic 版本 v.0.39
- PEAR 版本v.0.9.11 (Zhang et al., 2014)
- VSEARCH 版本 v2.13.7 (Rognes et al., 2016)
- BWA aligner 版本 v.0.7.12 (Li & Durbin, 2009)
- SAMTools 版本 v.1.7 (Li et al., 2009)
- perl在 CPAN modules 安装如下库: GetOpt::Long, IO::ZLib, List::Util, List::MoreUtils, Parallel::ForkManager
流程(共4块,7个步骤):
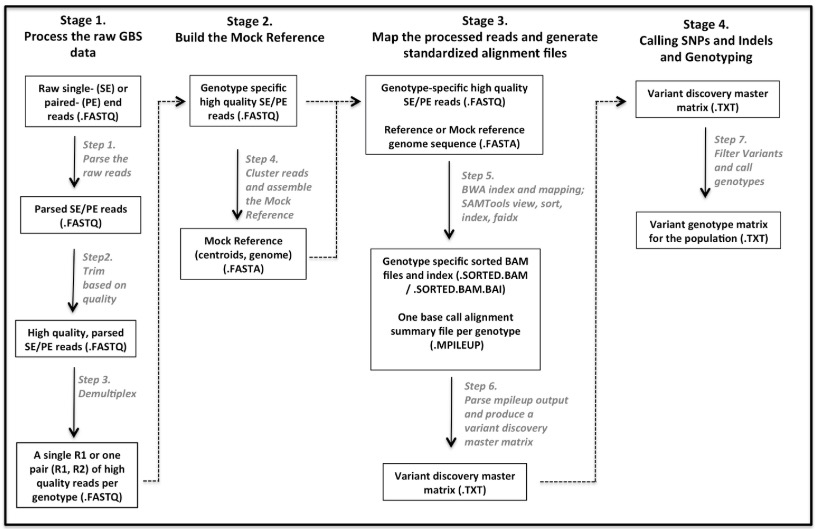
1. 处理原始raw GBS数据
- 步骤 1: 解析raw reads
- 步骤 2: 根据质量和接头进行过滤
- 步骤 3: Demultiplex(将multiplexed的reads根据index从不同或者同一个lane中分出,生成sample对应的fastq文件)
2. 建立 Mock Reference
- 步骤 4: 通过Reads聚类建立Mock Reference
3. 进行reads映射、标准比对
- 步骤 5: 用BWA-mem、SAMtools比对和 组装
- 步骤 6: 解析输出内容,生成variants矩阵
4. 基因组变异检测和鉴定出个体的每个变异位点的基因型
- 步骤 7: 过滤变异和鉴定出个体的每个变异位点的基因型
目前已经更新到v4.1版本。更多详细内容建议根据官方文档指导操作。
注意:目前v4.1版本有bug,可以查看pull了解
官方指导手册:https://github.com/halelab/GBS-SNP-CROP/wiki/GBS-SNP-CROP-User-Manual-(v.4.1)
参考资料:
1.https://github.com/halelab/GBS-SNP-CROP
2.Melo, A.T.O., Bartaula, R. & Hale, I. GBS-SNP-CROP: a reference-optional pipeline for SNP discovery and plant germplasm characterization using variable length, paired-end genotyping-by-sequencing data. BMC Bioinformatics17, 29 (2016).
3.https://www.jianshu.com/p/29fa856b83c2

浙江理工
已解决,是格式问题,将CR/LF格式统一转换成CR格式之后就可以运行,谢谢
浙江理工
您好好,我的程序在进行第八步,转换成VCF格式时出现问题:Can’t load GSC.GenoMatrix.txt file at /home/xujun/software/GBS-SNP-CROPv.4.1/GBS-SNP-CROP-8.pl line 258.您是否可以帮我看看,谢谢您!
陈浩
你输入的命令是什么?
陈浩
-in参数是必须的,怀疑你是没有提供这个参数和文件,然后的调用了默认的GSC.GenoMatrix.txt文件导致
大卫天龙
你好,我的步骤七中转换成VCF格式出现这样的错误:can’t load GSC.GenoMatrix48.txt file at GBS-SNP-CROPv.4.1/GBS-SNP-CROP-8.pl line 258.您可以帮我看看吗?谢谢!
陈浩
-in参数是必须的,怀疑你是没有提供这个参数和文件,然后的调用了默认的GSC.GenoMatrix.txt文件导致
2
你好,我的代码一直显示barcode failed,请问是怎么回事
陈浩
barcode文件是tab分割的文本文件,如下所示:
TGACGCCA Lib1_01 YES
CAGATA Lib1_02 YES
GAAGTG Lib2_05 YES
TAGCGGAT Lib3_10 NO
TATTCGCAT Lib3_11 YES
1
你好,我想请问第二阶段BarcodeID的使用,谢谢
陈浩
BarcodeID主要是和测序文件做匹配,格式如下(主要包含3列):
Barcode Genotype name Used for building the Mock Reference?
TGACGCCA Lib1_01 YES
CAGATA Lib1_02 YES
GAAGTG Lib2_05 YES
TAGCGGAT Lib3_10 NO
TATTCGCAT Lib3_11 YES
https://github.com/halelab/GBS-SNP-CROP/wiki/Appendix-A
2
你好,我的代码一直显示barcode failed,请问是怎么回事
陈浩
你是运行第几步显示barcode failed,你可以把你运行的命令贴上来。
陈浩
注意:目前最新流程部分脚本还是有点bug的,已经给官方提交了修改。
具体流程建议参考:https://github.com/halelab/GBS-SNP-CROP/wiki/GBS-SNP-CROP-User-Manual-(v.4.1)
但是流程文档部分参数可能有误,注意更新。