
GATK(Genome Analysis ToolKit),是目前最流行的基因组测序数据中分析变异信息的标准软件,也是目前最主流的call variant的软件之一。
目前最新版本文为4.1.9.0。 和之前的版本相比,GATK4在算法上进行了优化,运行速率有所提高,而且整合了picard 、Mutect2等一系列功能。关于GATK的介绍此处不在赘述(网络上介绍有很多)。

首先安装GATK4(需要java环境,1.8+版本):
Github下载页面:https://github.com/broadinstitute/gatk/releases
最新版下载:gatk-4.1.9.0.zip
Docker 镜像:https://hub.docker.com/r/broadinstitute/gatk/

下载ref参考数据库:
地址:ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/
GATK4 的官方教程给出了5套常用的pipeline:
- Germline SNPs + Indels
- Somatic SNVs + Indels
- RNAseq SNPs + Indels
- Germline CNVs
- Somatic CNVs
本文主要介绍下Germline SNPs + Indels和Somatic SNVs + Indels两个流程,其他流程如法炮制。其中Somatic突变是体细胞突变,主要侧重比较的是正常组织和患病组织的体细胞突变,一般来说是后天导致的疾病;Germline突变是可以遗传的生殖细胞突变,一般研究遗传性疾病。
1)Germline SNPs + Indels
分为Cohort数据分析步骤和单样本分析步骤
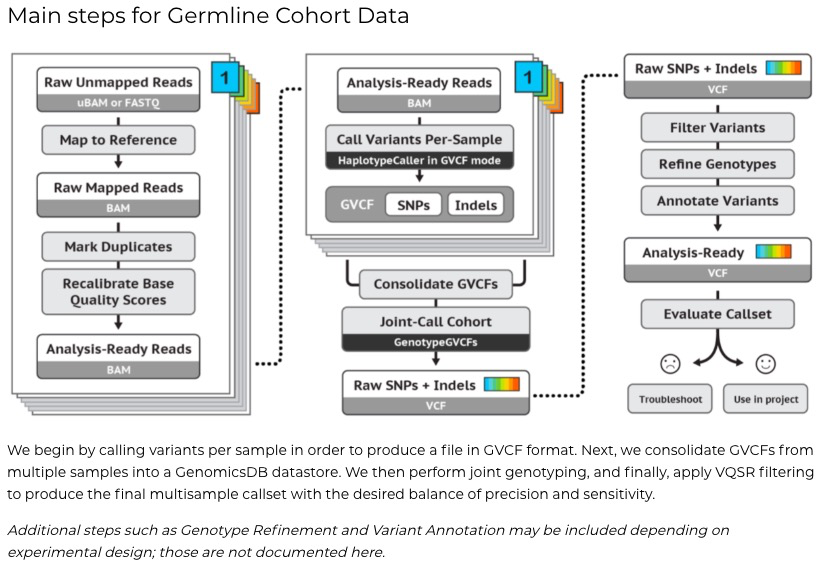

官方流程(基于CWL,推荐使用云端Cromwell来搭建):
## 脚本地址:https://github.com/gatk-workflows/gatk4-germline-snps-indels
version 1.0
## 官方介绍信息
## Copyright Broad Institute, 2019
##
## The haplotypecaller-gvcf-gatk4 workflow runs the HaplotypeCaller tool
## from GATK4 in GVCF mode on a single sample according to GATK Best Practices.
## When executed the workflow scatters the HaplotypeCaller tool over a sample
## using an intervals list file. The output file produced will be a
## single gvcf file which can be used by the joint-discovery workflow.
##
## Requirements/expectations :
## - One analysis-ready BAM file for a single sample (as identified in RG:SM)
## - Set of variant calling intervals lists for the scatter, provided in a file
##
## Outputs :
## - One GVCF file and its index
##
## Cromwell version support
## - Successfully tested on v53
##
## Runtime parameters are optimized for Broad's Google Cloud Platform implementation.
##
## LICENSING :
## This script is released under the WDL source code license (BSD-3) (see LICENSE in
## https://github.com/broadinstitute/wdl). Note however that the programs it calls may
## be subject to different licenses. Users are responsible for checking that they are
## authorized to run all programs before running this script. Please see the dockers
## for detailed licensing information pertaining to the included programs.
# WORKFLOW DEFINITION
workflow HaplotypeCallerGvcf_GATK4 {
input {
File input_bam
File input_bam_index
File ref_dict
File ref_fasta
File ref_fasta_index
File scattered_calling_intervals_list
Boolean make_gvcf = true
Boolean make_bamout = false
String gatk_docker = "us.gcr.io/broad-gatk/gatk:4.1.9.0"
String gatk_path = "/gatk/gatk"
String gitc_docker = "us.gcr.io/broad-gotc-prod/genomes-in-the-cloud:2.4.7-1603303710"
String samtools_path = "samtools"
}
Array[File] scattered_calling_intervals = read_lines(scattered_calling_intervals_list)
#is the input a cram file?
Boolean is_cram = sub(basename(input_bam), ".*\\.", "") == "cram"
String sample_basename = if is_cram then basename(input_bam, ".cram") else basename(input_bam, ".bam")
String vcf_basename = sample_basename
String output_suffix = if make_gvcf then ".g.vcf.gz" else ".vcf.gz"
String output_filename = vcf_basename + output_suffix
# We need disk to localize the sharded input and output due to the scatter for HaplotypeCaller.
# If we take the number we are scattering by and reduce by 20 we will have enough disk space
# to account for the fact that the data is quite uneven across the shards.
Int potential_hc_divisor = length(scattered_calling_intervals) - 20
Int hc_divisor = if potential_hc_divisor > 1 then potential_hc_divisor else 1
if ( is_cram ) {
call CramToBamTask {
input:
input_cram = input_bam,
sample_name = sample_basename,
ref_dict = ref_dict,
ref_fasta = ref_fasta,
ref_fasta_index = ref_fasta_index,
docker = gitc_docker,
samtools_path = samtools_path
}
}
# Call variants in parallel over grouped calling intervals
scatter (interval_file in scattered_calling_intervals) {
# Generate GVCF by interval
call HaplotypeCaller {
input:
input_bam = select_first([CramToBamTask.output_bam, input_bam]),
input_bam_index = select_first([CramToBamTask.output_bai, input_bam_index]),
interval_list = interval_file,
output_filename = output_filename,
ref_dict = ref_dict,
ref_fasta = ref_fasta,
ref_fasta_index = ref_fasta_index,
hc_scatter = hc_divisor,
make_gvcf = make_gvcf,
make_bamout = make_bamout,
docker = gatk_docker,
gatk_path = gatk_path
}
}
# Merge per-interval GVCFs
call MergeGVCFs {
input:
input_vcfs = HaplotypeCaller.output_vcf,
input_vcfs_indexes = HaplotypeCaller.output_vcf_index,
output_filename = output_filename,
docker = gatk_docker,
gatk_path = gatk_path
}
# Outputs that will be retained when execution is complete
output {
File output_vcf = MergeGVCFs.output_vcf
File output_vcf_index = MergeGVCFs.output_vcf_index
}
}
# TASK DEFINITIONS
task CramToBamTask {
input {
# Command parameters
File ref_fasta
File ref_fasta_index
File ref_dict
File input_cram
String sample_name
# Runtime parameters
String docker
Int? machine_mem_gb
Int? disk_space_gb
Boolean use_ssd = false
Int? preemptible_attempts
String samtools_path
}
Float output_bam_size = size(input_cram, "GB") / 0.60
Float ref_size = size(ref_fasta, "GB") + size(ref_fasta_index, "GB") + size(ref_dict, "GB")
Int disk_size = ceil(size(input_cram, "GB") + output_bam_size + ref_size) + 20
command {
set -e
set -o pipefail
~{samtools_path} view -h -T ~{ref_fasta} ~{input_cram} |
~{samtools_path} view -b -o ~{sample_name}.bam -
~{samtools_path} index -b ~{sample_name}.bam
mv ~{sample_name}.bam.bai ~{sample_name}.bai
}
runtime {
docker: docker
memory: select_first([machine_mem_gb, 15]) + " GB"
disks: "local-disk " + select_first([disk_space_gb, disk_size]) + if use_ssd then " SSD" else " HDD"
preemptible: select_first([preemptible_attempts, 3])
}
output {
File output_bam = "~{sample_name}.bam"
File output_bai = "~{sample_name}.bai"
}
}
# HaplotypeCaller per-sample in GVCF mode
task HaplotypeCaller {
input {
# Command parameters
File input_bam
File input_bam_index
File interval_list
String output_filename
File ref_dict
File ref_fasta
File ref_fasta_index
Float? contamination
Boolean make_gvcf
Boolean make_bamout
Int hc_scatter
String? gcs_project_for_requester_pays
String gatk_path
String? java_options
# Runtime parameters
String docker
Int? mem_gb
Int? disk_space_gb
Boolean use_ssd = false
Int? preemptible_attempts
}
String java_opt = select_first([java_options, "-XX:GCTimeLimit=50 -XX:GCHeapFreeLimit=10"])
Int machine_mem_gb = select_first([mem_gb, 7])
Int command_mem_gb = machine_mem_gb - 1
Float ref_size = size(ref_fasta, "GB") + size(ref_fasta_index, "GB") + size(ref_dict, "GB")
Int disk_size = ceil(((size(input_bam, "GB") + 30) / hc_scatter) + ref_size) + 20
String vcf_basename = if make_gvcf then basename(output_filename, ".gvcf") else basename(output_filename, ".vcf")
String bamout_arg = if make_bamout then "-bamout ~{vcf_basename}.bamout.bam" else ""
parameter_meta {
input_bam: {
description: "a bam file",
localization_optional: true
}
input_bam_index: {
description: "an index file for the bam input",
localization_optional: true
}
}
command {
set -e
~{gatk_path} --java-options "-Xmx~{command_mem_gb}G ~{java_opt}" \
HaplotypeCaller \
-R ~{ref_fasta} \
-I ~{input_bam} \
-L ~{interval_list} \
-O ~{output_filename} \
-contamination ~{default="0" contamination} \
-G StandardAnnotation -G StandardHCAnnotation ~{true="-G AS_StandardAnnotation" false="" make_gvcf} \
-GQB 10 -GQB 20 -GQB 30 -GQB 40 -GQB 50 -GQB 60 -GQB 70 -GQB 80 -GQB 90 \
~{true="-ERC GVCF" false="" make_gvcf} \
~{if defined(gcs_project_for_requester_pays) then "--gcs-project-for-requester-pays ~{gcs_project_for_requester_pays}" else ""} \
~{bamout_arg}
# Cromwell doesn't like optional task outputs, so we have to touch this file.
touch ~{vcf_basename}.bamout.bam
}
runtime {
docker: docker
memory: machine_mem_gb + " GB"
disks: "local-disk " + select_first([disk_space_gb, disk_size]) + if use_ssd then " SSD" else " HDD"
preemptible: select_first([preemptible_attempts, 3])
}
output {
File output_vcf = "~{output_filename}"
File output_vcf_index = "~{output_filename}.tbi"
File bamout = "~{vcf_basename}.bamout.bam"
}
}
# Merge GVCFs generated per-interval for the same sample
task MergeGVCFs {
input {
# Command parameters
Array[File] input_vcfs
Array[File] input_vcfs_indexes
String output_filename
String gatk_path
# Runtime parameters
String docker
Int? mem_gb
Int? disk_space_gb
Int? preemptible_attempts
}
Boolean use_ssd = false
Int machine_mem_gb = select_first([mem_gb, 3])
Int command_mem_gb = machine_mem_gb - 1
command {
set -e
~{gatk_path} --java-options "-Xmx~{command_mem_gb}G" \
MergeVcfs \
--INPUT ~{sep=' --INPUT ' input_vcfs} \
--OUTPUT ~{output_filename}
}
runtime {
docker: docker
memory: machine_mem_gb + " GB"
disks: "local-disk " + select_first([disk_space_gb, 100]) + if use_ssd then " SSD" else " HDD"
preemptible: select_first([preemptible_attempts, 3])
}
output {
File output_vcf = "~{output_filename}"
File output_vcf_index = "~{output_filename}.tbi"
}
}2)Somatic SNVs + Indels

## 脚本地址:https://github.com/gatk-workflows/gatk4-somatic-snvs-indels
version 1.0
## 官方介绍信息
## Copyright Broad Institute, 2017
##
## This WDL workflow runs GATK4 Mutect 2 on a single tumor-normal pair or on a single tumor sample,
## and performs additional filtering and functional annotation tasks.
##
## Main requirements/expectations :
## - One analysis-ready BAM file (and its index) for each sample
##
## Description of inputs:
##
## ** Runtime **
## gatk_docker: docker image to use for GATK 4 Mutect2
## preemptible: how many preemptions to tolerate before switching to a non-preemptible machine (on Google)
## max_retries: how many times to retry failed tasks -- very important on the cloud when there are transient errors
## gatk_override: (optional) local file or Google bucket path to a GATK 4 java jar file to be used instead of the GATK 4 jar
## in the docker image. This must be supplied when running in an environment that does not support docker
## (e.g. SGE cluster on a Broad on-prem VM)
##
## ** Workflow options **
## intervals: genomic intervals (will be used for scatter)
## scatter_count: number of parallel jobs to generate when scattering over intervals
## m2_extra_args, m2_extra_filtering_args: additional arguments for Mutect2 calling and filtering (optional)
## split_intervals_extra_args: additional arguments for splitting intervals before scattering (optional)
## run_orientation_bias_mixture_model_filter: (optional) if true, filter orientation bias sites with the read orientation artifact mixture model.
##
## ** Primary inputs **
## ref_fasta, ref_fai, ref_dict: reference genome, index, and dictionary
## tumor_bam, tumor_bam_index: BAM and index for the tumor sample
## normal_bam, normal_bam_index: BAM and index for the normal sample
##
## ** Primary resources ** (optional but strongly recommended)
## pon, pon_idx: optional panel of normals (and its index) in VCF format containing probable technical artifacts (false positves)
## gnomad, gnomad_idx: optional database of known germline variants (and its index) (see http://gnomad.broadinstitute.org/downloads)
## variants_for_contamination, variants_for_contamination_idx: VCF of common variants (and its index)with allele frequencies for calculating contamination
##
## ** Secondary resources ** (for optional tasks)
## realignment_index_bundle: resource for FilterAlignmentArtifacts, which runs if and only if it is specified. Generated by BwaMemIndexImageCreator.
##
## Funcotator parameters (see Funcotator help for more details).
## funco_reference_version: "hg19" for hg19 or b37. "hg38" for hg38. Default: "hg19"
## funco_output_format: "MAF" to produce a MAF file, "VCF" to procude a VCF file. Default: "MAF"
## funco_compress: (Only valid if funco_output_format == "VCF" ) If true, will compress the output of Funcotator. If false, produces an uncompressed output file. Default: false
## funco_use_gnomad_AF: If true, will include gnomAD allele frequency annotations in output by connecting to the internet to query gnomAD (this impacts performance). If false, will not annotate with gnomAD. Default: false
## funco_transcript_selection_mode: How to select transcripts in Funcotator. ALL, CANONICAL, or BEST_EFFECT
## funco_transcript_selection_list: Transcripts (one GENCODE ID per line) to give priority during selection process.
## funco_data_sources_tar_gz: Funcotator datasources tar gz file. Bucket location is recommended when running on the cloud.
## funco_annotation_defaults: Default values for annotations, when values are unspecified. Specified as <ANNOTATION>:<VALUE>. For example: "Center:Broad"
## funco_annotation_overrides: Values for annotations, even when values are unspecified. Specified as <ANNOTATION>:<VALUE>. For example: "Center:Broad"
## funcotator_excluded_fields: Annotations that should not appear in the output (VCF or MAF). Specified as <ANNOTATION>. For example: "ClinVar_ALLELEID"
## funco_filter_funcotations: If true, will only annotate variants that have passed filtering (. or PASS value in the FILTER column). If false, will annotate all variants in the input file. Default: true
## funcotator_extra_args: Any additional arguments to pass to Funcotator. Default: ""
##
## Outputs :
## - One VCF file and its index with primary filtering applied; secondary filtering and functional annotation if requested; a bamout.bam
## file of reassembled reads if requested
##
## Cromwell version support
## - Successfully tested on v34
##
## LICENSING :
## This script is released under the WDL source code license (BSD-3) (see LICENSE in
## https://github.com/broadinstitute/wdl). Note however that the programs it calls may
## be subject to different licenses. Users are responsible for checking that they are
## authorized to run all programs before running this script. Please see the docker
## pages at https://hub.docker.com/r/broadinstitute/* for detailed licensing information
## pertaining to the included programs.
struct Runtime {
String gatk_docker
File? gatk_override
Int max_retries
Int preemptible
Int cpu
Int machine_mem
Int command_mem
Int disk
Int boot_disk_size
}
workflow Mutect2 {
input {
# Mutect2 inputs
File? intervals
File ref_fasta
File ref_fai
File ref_dict
File tumor_reads
File tumor_reads_index
File? normal_reads
File? normal_reads_index
File? pon
File? pon_idx
Int scatter_count
File? gnomad
File? gnomad_idx
File? variants_for_contamination
File? variants_for_contamination_idx
File? realignment_index_bundle
String? realignment_extra_args
Boolean? run_orientation_bias_mixture_model_filter
String? m2_extra_args
String? m2_extra_filtering_args
String? split_intervals_extra_args
Boolean? make_bamout
Boolean? compress_vcfs
File? gga_vcf
File? gga_vcf_idx
# Funcotator inputs
Boolean? run_funcotator
String? sequencing_center
String? sequence_source
String? funco_reference_version
String? funco_output_format
Boolean? funco_compress
Boolean? funco_use_gnomad_AF
File? funco_data_sources_tar_gz
String? funco_transcript_selection_mode
File? funco_transcript_selection_list
Array[String]? funco_annotation_defaults
Array[String]? funco_annotation_overrides
Array[String]? funcotator_excluded_fields
Boolean? funco_filter_funcotations
String? funcotator_extra_args
String funco_default_output_format = "MAF"
# runtime
String gatk_docker
File? gatk_override
String basic_bash_docker = "ubuntu:16.04"
Boolean? filter_funcotations
Int? preemptible
Int? max_retries
Int small_task_cpu = 2
Int small_task_mem = 4
Int small_task_disk = 100
Int boot_disk_size = 12
Int learn_read_orientation_mem = 8000
Int filter_alignment_artifacts_mem = 9000
# Use as a last resort to increase the disk given to every task in case of ill behaving data
Int? emergency_extra_disk
# These are multipliers to multipler inputs by to make sure we have enough disk to accommodate for possible output sizes
# Large is for Bams/WGS vcfs
# Small is for metrics/other vcfs
Float large_input_to_output_multiplier = 2.25
Float small_input_to_output_multiplier = 2.0
Float cram_to_bam_multiplier = 6.0
}
Int preemptible_or_default = select_first([preemptible, 2])
Int max_retries_or_default = select_first([max_retries, 2])
Boolean compress = select_first([compress_vcfs, false])
Boolean run_ob_filter = select_first([run_orientation_bias_mixture_model_filter, false])
Boolean make_bamout_or_default = select_first([make_bamout, false])
Boolean run_funcotator_or_default = select_first([run_funcotator, false])
Boolean filter_funcotations_or_default = select_first([filter_funcotations, true])
# Disk sizes used for dynamic sizing
Int ref_size = ceil(size(ref_fasta, "GB") + size(ref_dict, "GB") + size(ref_fai, "GB"))
Int tumor_reads_size = ceil(size(tumor_reads, "GB") + size(tumor_reads_index, "GB"))
Int gnomad_vcf_size = if defined(gnomad) then ceil(size(gnomad, "GB")) else 0
Int normal_reads_size = if defined(normal_reads) then ceil(size(normal_reads, "GB") + size(normal_reads_index, "GB")) else 0
# If no tar is provided, the task downloads one from broads ftp server
Int funco_tar_size = if defined(funco_data_sources_tar_gz) then ceil(size(funco_data_sources_tar_gz, "GB") * 3) else 100
Int gatk_override_size = if defined(gatk_override) then ceil(size(gatk_override, "GB")) else 0
# This is added to every task as padding, should increase if systematically you need more disk for every call
Int disk_pad = 10 + gatk_override_size + select_first([emergency_extra_disk,0])
# logic about output file names -- these are the names *without* .vcf extensions
String output_basename = basename(basename(tumor_reads, ".bam"),".cram") #hacky way to strip either .bam or .cram
String unfiltered_name = output_basename + "-unfiltered"
String filtered_name = output_basename + "-filtered"
String funcotated_name = output_basename + "-funcotated"
String output_vcf_name = output_basename + ".vcf"
Int tumor_cram_to_bam_disk = ceil(tumor_reads_size * cram_to_bam_multiplier)
Int normal_cram_to_bam_disk = ceil(normal_reads_size * cram_to_bam_multiplier)
Runtime standard_runtime = {"gatk_docker": gatk_docker, "gatk_override": gatk_override,
"max_retries": max_retries_or_default, "preemptible": preemptible_or_default, "cpu": small_task_cpu,
"machine_mem": small_task_mem * 1000, "command_mem": small_task_mem * 1000 - 500,
"disk": small_task_disk + disk_pad, "boot_disk_size": boot_disk_size}
if (basename(tumor_reads) != basename(tumor_reads, ".cram")) {
call CramToBam as TumorCramToBam {
input:
ref_fasta = ref_fasta,
ref_fai = ref_fai,
ref_dict = ref_dict,
cram = tumor_reads,
crai = tumor_reads_index,
name = output_basename,
disk_size = tumor_cram_to_bam_disk
}
}
String normal_or_empty = select_first([normal_reads, ""])
if (basename(normal_or_empty) != basename(normal_or_empty, ".cram")) {
String normal_basename = basename(basename(normal_or_empty, ".bam"),".cram")
call CramToBam as NormalCramToBam {
input:
ref_fasta = ref_fasta,
ref_fai = ref_fai,
ref_dict = ref_dict,
cram = normal_reads,
crai = normal_reads_index,
name = normal_basename,
disk_size = normal_cram_to_bam_disk
}
}
File tumor_bam = select_first([TumorCramToBam.output_bam, tumor_reads])
File tumor_bai = select_first([TumorCramToBam.output_bai, tumor_reads_index])
File? normal_bam = if defined(normal_reads) then select_first([NormalCramToBam.output_bam, normal_reads]) else normal_reads
File? normal_bai = if defined(normal_reads) then select_first([NormalCramToBam.output_bai, normal_reads_index]) else normal_reads_index
Int tumor_bam_size = ceil(size(tumor_bam, "GB") + size(tumor_bai, "GB"))
Int normal_bam_size = if defined(normal_bam) then ceil(size(normal_bam, "GB") + size(normal_bai, "GB")) else 0
Int m2_output_size = tumor_bam_size / scatter_count
#TODO: do we need to change this disk size now that NIO is always going to happen (for the google backend only)
Int m2_per_scatter_size = (tumor_bam_size + normal_bam_size) + ref_size + gnomad_vcf_size + m2_output_size + disk_pad
call SplitIntervals {
input:
intervals = intervals,
ref_fasta = ref_fasta,
ref_fai = ref_fai,
ref_dict = ref_dict,
scatter_count = scatter_count,
split_intervals_extra_args = split_intervals_extra_args,
runtime_params = standard_runtime
}
scatter (subintervals in SplitIntervals.interval_files ) {
call M2 {
input:
intervals = subintervals,
ref_fasta = ref_fasta,
ref_fai = ref_fai,
ref_dict = ref_dict,
tumor_bam = tumor_bam,
tumor_bai = tumor_bai,
normal_bam = normal_bam,
normal_bai = normal_bai,
pon = pon,
pon_idx = pon_idx,
gnomad = gnomad,
gnomad_idx = gnomad_idx,
preemptible = preemptible,
max_retries = max_retries,
m2_extra_args = m2_extra_args,
variants_for_contamination = variants_for_contamination,
variants_for_contamination_idx = variants_for_contamination_idx,
make_bamout = make_bamout_or_default,
run_ob_filter = run_ob_filter,
compress = compress,
gga_vcf = gga_vcf,
gga_vcf_idx = gga_vcf_idx,
gatk_override = gatk_override,
gatk_docker = gatk_docker,
disk_space = m2_per_scatter_size
}
}
Int merged_vcf_size = ceil(size(M2.unfiltered_vcf, "GB"))
Int merged_bamout_size = ceil(size(M2.output_bamOut, "GB"))
if (run_ob_filter) {
call LearnReadOrientationModel {
input:
f1r2_tar_gz = M2.f1r2_counts,
runtime_params = standard_runtime,
mem = learn_read_orientation_mem
}
}
call MergeVCFs {
input:
input_vcfs = M2.unfiltered_vcf,
input_vcf_indices = M2.unfiltered_vcf_idx,
output_name = unfiltered_name,
compress = compress,
runtime_params = standard_runtime
}
if (make_bamout_or_default) {
call MergeBamOuts {
input:
ref_fasta = ref_fasta,
ref_fai = ref_fai,
ref_dict = ref_dict,
bam_outs = M2.output_bamOut,
output_vcf_name = basename(MergeVCFs.merged_vcf, ".vcf"),
runtime_params = standard_runtime,
disk_space = ceil(merged_bamout_size * large_input_to_output_multiplier) + disk_pad,
}
}
call MergeStats { input: stats = M2.stats, runtime_params = standard_runtime }
if (defined(variants_for_contamination)) {
call MergePileupSummaries as MergeTumorPileups {
input:
input_tables = flatten(M2.tumor_pileups),
output_name = output_basename,
ref_dict = ref_dict,
runtime_params = standard_runtime
}
if (defined(normal_bam)){
call MergePileupSummaries as MergeNormalPileups {
input:
input_tables = flatten(M2.normal_pileups),
output_name = output_basename,
ref_dict = ref_dict,
runtime_params = standard_runtime
}
}
call CalculateContamination {
input:
tumor_pileups = MergeTumorPileups.merged_table,
normal_pileups = MergeNormalPileups.merged_table,
runtime_params = standard_runtime
}
}
call Filter {
input:
ref_fasta = ref_fasta,
ref_fai = ref_fai,
ref_dict = ref_dict,
intervals = intervals,
unfiltered_vcf = MergeVCFs.merged_vcf,
unfiltered_vcf_idx = MergeVCFs.merged_vcf_idx,
output_name = filtered_name,
compress = compress,
mutect_stats = MergeStats.merged_stats,
contamination_table = CalculateContamination.contamination_table,
maf_segments = CalculateContamination.maf_segments,
artifact_priors_tar_gz = LearnReadOrientationModel.artifact_prior_table,
m2_extra_filtering_args = m2_extra_filtering_args,
runtime_params = standard_runtime,
disk_space = ceil(size(MergeVCFs.merged_vcf, "GB") * small_input_to_output_multiplier) + disk_pad
}
if (defined(realignment_index_bundle)) {
call FilterAlignmentArtifacts {
input:
bam = tumor_bam,
bai = tumor_bai,
realignment_index_bundle = select_first([realignment_index_bundle]),
realignment_extra_args = realignment_extra_args,
compress = compress,
output_name = filtered_name,
input_vcf = Filter.filtered_vcf,
input_vcf_idx = Filter.filtered_vcf_idx,
runtime_params = standard_runtime,
mem = filter_alignment_artifacts_mem
}
}
if (run_funcotator_or_default) {
File funcotate_vcf_input = select_first([FilterAlignmentArtifacts.filtered_vcf, Filter.filtered_vcf])
File funcotate_vcf_input_index = select_first([FilterAlignmentArtifacts.filtered_vcf_idx, Filter.filtered_vcf_idx])
call Funcotate {
input:
ref_fasta = ref_fasta,
ref_fai = ref_fai,
ref_dict = ref_dict,
input_vcf = funcotate_vcf_input,
input_vcf_idx = funcotate_vcf_input_index,
reference_version = select_first([funco_reference_version, "hg19"]),
output_file_base_name = basename(funcotate_vcf_input, ".vcf") + ".annotated",
output_format = if defined(funco_output_format) then "" + funco_output_format else funco_default_output_format,
compress = if defined(funco_compress) then select_first([funco_compress]) else false,
use_gnomad = if defined(funco_use_gnomad_AF) then select_first([funco_use_gnomad_AF]) else false,
data_sources_tar_gz = funco_data_sources_tar_gz,
case_id = M2.tumor_sample[0],
control_id = M2.normal_sample[0],
sequencing_center = sequencing_center,
sequence_source = sequence_source,
transcript_selection_mode = funco_transcript_selection_mode,
transcript_selection_list = funco_transcript_selection_list,
annotation_defaults = funco_annotation_defaults,
annotation_overrides = funco_annotation_overrides,
funcotator_excluded_fields = funcotator_excluded_fields,
filter_funcotations = filter_funcotations_or_default,
extra_args = funcotator_extra_args,
runtime_params = standard_runtime,
disk_space = ceil(size(funcotate_vcf_input, "GB") * large_input_to_output_multiplier) + funco_tar_size + disk_pad
}
}
output {
File filtered_vcf = select_first([FilterAlignmentArtifacts.filtered_vcf, Filter.filtered_vcf])
File filtered_vcf_idx = select_first([FilterAlignmentArtifacts.filtered_vcf_idx, Filter.filtered_vcf_idx])
File filtering_stats = Filter.filtering_stats
File mutect_stats = MergeStats.merged_stats
File? contamination_table = CalculateContamination.contamination_table
File? funcotated_file = Funcotate.funcotated_output_file
File? funcotated_file_index = Funcotate.funcotated_output_file_index
File? bamout = MergeBamOuts.merged_bam_out
File? bamout_index = MergeBamOuts.merged_bam_out_index
File? maf_segments = CalculateContamination.maf_segments
File? read_orientation_model_params = LearnReadOrientationModel.artifact_prior_table
}
}
task CramToBam {
input {
File ref_fasta
File ref_fai
File ref_dict
#cram and crai must be optional since Normal cram is optional
File? cram
File? crai
String name
Int disk_size
Int? mem
}
Int machine_mem = if defined(mem) then mem * 1000 else 6000
#Calls samtools view to do the conversion
command {
#Set -e and -o says if any command I run fails in this script, make sure to return a failure
set -e
set -o pipefail
samtools view -h -T ~{ref_fasta} ~{cram} |
samtools view -b -o ~{name}.bam -
samtools index -b ~{name}.bam
mv ~{name}.bam.bai ~{name}.bai
}
runtime {
docker: "us.gcr.io/broad-gotc-prod/genomes-in-the-cloud:2.3.3-1513176735"
memory: machine_mem + " MB"
disks: "local-disk " + disk_size + " HDD"
}
output {
File output_bam = "~{name}.bam"
File output_bai = "~{name}.bai"
}
}
task SplitIntervals {
input {
File? intervals
File ref_fasta
File ref_fai
File ref_dict
Int scatter_count
String? split_intervals_extra_args
# runtime
Runtime runtime_params
}
command {
set -e
export GATK_LOCAL_JAR=~{default="/root/gatk.jar" runtime_params.gatk_override}
mkdir interval-files
gatk --java-options "-Xmx~{runtime_params.command_mem}m" SplitIntervals \
-R ~{ref_fasta} \
~{"-L " + intervals} \
-scatter ~{scatter_count} \
-O interval-files \
~{split_intervals_extra_args}
cp interval-files/*.interval_list .
}
runtime {
docker: runtime_params.gatk_docker
bootDiskSizeGb: runtime_params.boot_disk_size
memory: runtime_params.machine_mem + " MB"
disks: "local-disk " + runtime_params.disk + " HDD"
preemptible: runtime_params.preemptible
maxRetries: runtime_params.max_retries
cpu: runtime_params.cpu
}
output {
Array[File] interval_files = glob("*.interval_list")
}
}
task M2 {
input {
File? intervals
File ref_fasta
File ref_fai
File ref_dict
File tumor_bam
File tumor_bai
File? normal_bam
File? normal_bai
File? pon
File? pon_idx
File? gnomad
File? gnomad_idx
String? m2_extra_args
Boolean? make_bamout
Boolean? run_ob_filter
Boolean compress
File? gga_vcf
File? gga_vcf_idx
File? variants_for_contamination
File? variants_for_contamination_idx
File? gatk_override
# runtime
String gatk_docker
Int? mem
Int? preemptible
Int? max_retries
Int? disk_space
Int? cpu
Boolean use_ssd = false
}
String output_vcf = "output" + if compress then ".vcf.gz" else ".vcf"
String output_vcf_idx = output_vcf + if compress then ".tbi" else ".idx"
String output_stats = output_vcf + ".stats"
# Mem is in units of GB but our command and memory runtime values are in MB
Int machine_mem = if defined(mem) then mem * 1000 else 3500
Int command_mem = machine_mem - 500
parameter_meta{
intervals: {localization_optional: true}
ref_fasta: {localization_optional: true}
ref_fai: {localization_optional: true}
ref_dict: {localization_optional: true}
tumor_bam: {localization_optional: true}
tumor_bai: {localization_optional: true}
normal_bam: {localization_optional: true}
normal_bai: {localization_optional: true}
pon: {localization_optional: true}
pon_idx: {localization_optional: true}
gnomad: {localization_optional: true}
gnomad_idx: {localization_optional: true}
gga_vcf: {localization_optional: true}
gga_vcf_idx: {localization_optional: true}
variants_for_contamination: {localization_optional: true}
variants_for_contamination_idx: {localization_optional: true}
}
command <<<
set -e
export GATK_LOCAL_JAR=~{default="/root/gatk.jar" gatk_override}
# We need to create these files regardless, even if they stay empty
touch bamout.bam
touch f1r2.tar.gz
echo "" > normal_name.txt
gatk --java-options "-Xmx~{command_mem}m" GetSampleName -R ~{ref_fasta} -I ~{tumor_bam} -O tumor_name.txt -encode
tumor_command_line="-I ~{tumor_bam} -tumor `cat tumor_name.txt`"
if [[ ! -z "~{normal_bam}" ]]; then
gatk --java-options "-Xmx~{command_mem}m" GetSampleName -R ~{ref_fasta} -I ~{normal_bam} -O normal_name.txt -encode
normal_command_line="-I ~{normal_bam} -normal `cat normal_name.txt`"
fi
gatk --java-options "-Xmx~{command_mem}m" Mutect2 \
-R ~{ref_fasta} \
$tumor_command_line \
$normal_command_line \
~{"--germline-resource " + gnomad} \
~{"-pon " + pon} \
~{"-L " + intervals} \
~{"--alleles " + gga_vcf} \
-O "~{output_vcf}" \
~{true='--bam-output bamout.bam' false='' make_bamout} \
~{true='--f1r2-tar-gz f1r2.tar.gz' false='' run_ob_filter} \
~{m2_extra_args}
m2_exit_code=$?
### GetPileupSummaries
# If the variants for contamination and the intervals for this scatter don't intersect, GetPileupSummaries
# throws an error. However, there is nothing wrong with an empty intersection for our purposes; it simply doesn't
# contribute to the merged pileup summaries that we create downstream. We implement this by with array outputs.
# If the tool errors, no table is created and the glob yields an empty array.
set +e
if [[ ! -z "~{variants_for_contamination}" ]]; then
gatk --java-options "-Xmx~{command_mem}m" GetPileupSummaries -R ~{ref_fasta} -I ~{tumor_bam} ~{"--interval-set-rule INTERSECTION -L " + intervals} \
-V ~{variants_for_contamination} -L ~{variants_for_contamination} -O tumor-pileups.table
if [[ ! -z "~{normal_bam}" ]]; then
gatk --java-options "-Xmx~{command_mem}m" GetPileupSummaries -R ~{ref_fasta} -I ~{normal_bam} ~{"--interval-set-rule INTERSECTION -L " + intervals} \
-V ~{variants_for_contamination} -L ~{variants_for_contamination} -O normal-pileups.table
fi
fi
# the script only fails if Mutect2 itself fails
exit $m2_exit_code
>>>
runtime {
docker: gatk_docker
bootDiskSizeGb: 12
memory: machine_mem + " MB"
disks: "local-disk " + select_first([disk_space, 100]) + if use_ssd then " SSD" else " HDD"
preemptible: select_first([preemptible, 10])
maxRetries: select_first([max_retries, 0])
cpu: select_first([cpu, 1])
}
output {
File unfiltered_vcf = "~{output_vcf}"
File unfiltered_vcf_idx = "~{output_vcf_idx}"
File output_bamOut = "bamout.bam"
String tumor_sample = read_string("tumor_name.txt")
String normal_sample = read_string("normal_name.txt")
File stats = "~{output_stats}"
File f1r2_counts = "f1r2.tar.gz"
Array[File] tumor_pileups = glob("*tumor-pileups.table")
Array[File] normal_pileups = glob("*normal-pileups.table")
}
}
task MergeVCFs {
input {
Array[File] input_vcfs
Array[File] input_vcf_indices
String output_name
Boolean compress
Runtime runtime_params
}
String output_vcf = output_name + if compress then ".vcf.gz" else ".vcf"
String output_vcf_idx = output_vcf + if compress then ".tbi" else ".idx"
# using MergeVcfs instead of GatherVcfs so we can create indices
# WARNING 2015-10-28 15:01:48 GatherVcfs Index creation not currently supported when gathering block compressed VCFs.
command {
set -e
export GATK_LOCAL_JAR=~{default="/root/gatk.jar" runtime_params.gatk_override}
gatk --java-options "-Xmx~{runtime_params.command_mem}m" MergeVcfs -I ~{sep=' -I ' input_vcfs} -O ~{output_vcf}
}
runtime {
docker: runtime_params.gatk_docker
bootDiskSizeGb: runtime_params.boot_disk_size
memory: runtime_params.machine_mem + " MB"
disks: "local-disk " + runtime_params.disk + " HDD"
preemptible: runtime_params.preemptible
maxRetries: runtime_params.max_retries
cpu: runtime_params.cpu
}
output {
File merged_vcf = "~{output_vcf}"
File merged_vcf_idx = "~{output_vcf_idx}"
}
}
task MergeBamOuts {
input {
File ref_fasta
File ref_fai
File ref_dict
Array[File]+ bam_outs
String output_vcf_name
Runtime runtime_params
Int? disk_space #override to request more disk than default small task params
}
command <<<
# This command block assumes that there is at least one file in bam_outs.
# Do not call this task if len(bam_outs) == 0
set -e
export GATK_LOCAL_JAR=~{default="/root/gatk.jar" runtime_params.gatk_override}
gatk --java-options "-Xmx~{runtime_params.command_mem}m" GatherBamFiles \
-I ~{sep=" -I " bam_outs} -O unsorted.out.bam -R ~{ref_fasta}
# We must sort because adjacent scatters may have overlapping (padded) assembly regions, hence
# overlapping bamouts
gatk --java-options "-Xmx~{runtime_params.command_mem}m" SortSam -I unsorted.out.bam \
-O ~{output_vcf_name}.out.bam \
--SORT_ORDER coordinate -VALIDATION_STRINGENCY LENIENT
gatk --java-options "-Xmx~{runtime_params.command_mem}m" BuildBamIndex -I ~{output_vcf_name}.out.bam -VALIDATION_STRINGENCY LENIENT
>>>
runtime {
docker: runtime_params.gatk_docker
bootDiskSizeGb: runtime_params.boot_disk_size
memory: runtime_params.machine_mem + " MB"
disks: "local-disk " + select_first([disk_space, runtime_params.disk]) + " HDD"
preemptible: runtime_params.preemptible
maxRetries: runtime_params.max_retries
cpu: runtime_params.cpu
}
output {
File merged_bam_out = "~{output_vcf_name}.out.bam"
File merged_bam_out_index = "~{output_vcf_name}.out.bai"
}
}
task MergeStats {
input {
Array[File]+ stats
Runtime runtime_params
}
command {
set -e
export GATK_LOCAL_JAR=~{default="/root/gatk.jar" runtime_params.gatk_override}
gatk --java-options "-Xmx~{runtime_params.command_mem}m" MergeMutectStats \
-stats ~{sep=" -stats " stats} -O merged.stats
}
runtime {
docker: runtime_params.gatk_docker
bootDiskSizeGb: runtime_params.boot_disk_size
memory: runtime_params.machine_mem + " MB"
disks: "local-disk " + runtime_params.disk + " HDD"
preemptible: runtime_params.preemptible
maxRetries: runtime_params.max_retries
cpu: runtime_params.cpu
}
output {
File merged_stats = "merged.stats"
}
}
task MergePileupSummaries {
input {
Array[File] input_tables
String output_name
File ref_dict
Runtime runtime_params
}
command {
set -e
export GATK_LOCAL_JAR=~{default="/root/gatk.jar" runtime_params.gatk_override}
gatk --java-options "-Xmx~{runtime_params.command_mem}m" GatherPileupSummaries \
--sequence-dictionary ~{ref_dict} \
-I ~{sep=' -I ' input_tables} \
-O ~{output_name}.tsv
}
runtime {
docker: runtime_params.gatk_docker
bootDiskSizeGb: runtime_params.boot_disk_size
memory: runtime_params.machine_mem + " MB"
disks: "local-disk " + runtime_params.disk + " HDD"
preemptible: runtime_params.preemptible
maxRetries: runtime_params.max_retries
cpu: runtime_params.cpu
}
output {
File merged_table = "~{output_name}.tsv"
}
}
# Learning step of the orientation bias mixture model, which is the recommended orientation bias filter as of September 2018
task LearnReadOrientationModel {
input {
Array[File] f1r2_tar_gz
Runtime runtime_params
Int? mem #override memory
}
Int machine_mem = select_first([mem, runtime_params.machine_mem])
Int command_mem = machine_mem - 1000
command {
set -e
export GATK_LOCAL_JAR=~{default="/root/gatk.jar" runtime_params.gatk_override}
gatk --java-options "-Xmx~{command_mem}m" LearnReadOrientationModel \
-I ~{sep=" -I " f1r2_tar_gz} \
-O "artifact-priors.tar.gz"
}
runtime {
docker: runtime_params.gatk_docker
bootDiskSizeGb: runtime_params.boot_disk_size
memory: machine_mem + " MB"
disks: "local-disk " + runtime_params.disk + " HDD"
preemptible: runtime_params.preemptible
maxRetries: runtime_params.max_retries
cpu: runtime_params.cpu
}
output {
File artifact_prior_table = "artifact-priors.tar.gz"
}
}
task CalculateContamination {
input {
String? intervals
File tumor_pileups
File? normal_pileups
Runtime runtime_params
}
command {
set -e
export GATK_LOCAL_JAR=~{default="/root/gatk.jar" runtime_params.gatk_override}
gatk --java-options "-Xmx~{runtime_params.command_mem}m" CalculateContamination -I ~{tumor_pileups} \
-O contamination.table --tumor-segmentation segments.table ~{"-matched " + normal_pileups}
}
runtime {
docker: runtime_params.gatk_docker
bootDiskSizeGb: runtime_params.boot_disk_size
memory: runtime_params.machine_mem + " MB"
disks: "local-disk " + runtime_params.disk + " HDD"
preemptible: runtime_params.preemptible
maxRetries: runtime_params.max_retries
cpu: runtime_params.cpu
}
output {
File contamination_table = "contamination.table"
File maf_segments = "segments.table"
}
}
task Filter {
input {
File? intervals
File ref_fasta
File ref_fai
File ref_dict
File unfiltered_vcf
File unfiltered_vcf_idx
String output_name
Boolean compress
File? mutect_stats
File? artifact_priors_tar_gz
File? contamination_table
File? maf_segments
String? m2_extra_filtering_args
Runtime runtime_params
Int? disk_space
}
String output_vcf = output_name + if compress then ".vcf.gz" else ".vcf"
String output_vcf_idx = output_vcf + if compress then ".tbi" else ".idx"
parameter_meta{
ref_fasta: {localization_optional: true}
ref_fai: {localization_optional: true}
ref_dict: {localization_optional: true}
}
command {
set -e
export GATK_LOCAL_JAR=~{default="/root/gatk.jar" runtime_params.gatk_override}
gatk --java-options "-Xmx~{runtime_params.command_mem}m" FilterMutectCalls -V ~{unfiltered_vcf} \
-R ~{ref_fasta} \
-O ~{output_vcf} \
~{"--contamination-table " + contamination_table} \
~{"--tumor-segmentation " + maf_segments} \
~{"--ob-priors " + artifact_priors_tar_gz} \
~{"-stats " + mutect_stats} \
--filtering-stats filtering.stats \
~{m2_extra_filtering_args}
}
runtime {
docker: runtime_params.gatk_docker
bootDiskSizeGb: runtime_params.boot_disk_size
memory: runtime_params.machine_mem + " MB"
disks: "local-disk " + select_first([disk_space, runtime_params.disk]) + " HDD"
preemptible: runtime_params.preemptible
maxRetries: runtime_params.max_retries
cpu: runtime_params.cpu
}
output {
File filtered_vcf = "~{output_vcf}"
File filtered_vcf_idx = "~{output_vcf_idx}"
File filtering_stats = "filtering.stats"
}
}
task FilterAlignmentArtifacts {
input {
File input_vcf
File input_vcf_idx
File bam
File bai
String output_name
Boolean compress
File realignment_index_bundle
String? realignment_extra_args
Runtime runtime_params
Int mem
}
String output_vcf = output_name + if compress then ".vcf.gz" else ".vcf"
String output_vcf_idx = output_vcf + if compress then ".tbi" else ".idx"
Int machine_mem = mem
Int command_mem = machine_mem - 500
parameter_meta{
input_vcf: {localization_optional: true}
input_vcf_idx: {localization_optional: true}
bam: {localization_optional: true}
bai: {localization_optional: true}
}
command {
set -e
export GATK_LOCAL_JAR=~{default="/root/gatk.jar" runtime_params.gatk_override}
gatk --java-options "-Xmx~{command_mem}m" FilterAlignmentArtifacts \
-V ~{input_vcf} \
-I ~{bam} \
--bwa-mem-index-image ~{realignment_index_bundle} \
~{realignment_extra_args} \
-O ~{output_vcf}
}
runtime {
docker: runtime_params.gatk_docker
bootDiskSizeGb: runtime_params.boot_disk_size
memory: machine_mem + " MB"
disks: "local-disk " + runtime_params.disk + " HDD"
preemptible: runtime_params.preemptible
maxRetries: runtime_params.max_retries
cpu: runtime_params.cpu
}
output {
File filtered_vcf = "~{output_vcf}"
File filtered_vcf_idx = "~{output_vcf_idx}"
}
}
task Funcotate {
input {
File ref_fasta
File ref_fai
File ref_dict
File input_vcf
File input_vcf_idx
String reference_version
String output_file_base_name
String output_format
Boolean compress
Boolean use_gnomad
# This should be updated when a new version of the data sources is released
# TODO: Make this dynamically chosen in the command.
File? data_sources_tar_gz = "gs://broad-public-datasets/funcotator/funcotator_dataSources.v1.6.20190124s.tar.gz"
String? control_id
String? case_id
String? sequencing_center
String? sequence_source
String? transcript_selection_mode
File? transcript_selection_list
Array[String]? annotation_defaults
Array[String]? annotation_overrides
Array[String]? funcotator_excluded_fields
Boolean? filter_funcotations
File? interval_list
String? extra_args
# ==============
Runtime runtime_params
Int? disk_space #override to request more disk than default small task params
# You may have to change the following two parameter values depending on the task requirements
Int default_ram_mb = 3000
# WARNING: In the workflow, you should calculate the disk space as an input to this task (disk_space_gb). Please see [TODO: Link from Jose] for examples.
Int default_disk_space_gb = 100
}
# ==============
# Process input args:
String output_maf = output_file_base_name + ".maf"
String output_maf_index = output_maf + ".idx"
String output_vcf = output_file_base_name + if compress then ".vcf.gz" else ".vcf"
String output_vcf_idx = output_vcf + if compress then ".tbi" else ".idx"
String output_file = if output_format == "MAF" then output_maf else output_vcf
String output_file_index = if output_format == "MAF" then output_maf_index else output_vcf_idx
String transcript_selection_arg = if defined(transcript_selection_list) then " --transcript-list " else ""
String annotation_def_arg = if defined(annotation_defaults) then " --annotation-default " else ""
String annotation_over_arg = if defined(annotation_overrides) then " --annotation-override " else ""
String filter_funcotations_args = if defined(filter_funcotations) && (filter_funcotations) then " --remove-filtered-variants " else ""
String excluded_fields_args = if defined(funcotator_excluded_fields) then " --exclude-field " else ""
String interval_list_arg = if defined(interval_list) then " -L " else ""
String extra_args_arg = select_first([extra_args, ""])
String dollar = "$"
parameter_meta{
ref_fasta: {localization_optional: true}
ref_fai: {localization_optional: true}
ref_dict: {localization_optional: true}
input_vcf: {localization_optional: true}
input_vcf_idx: {localization_optional: true}
}
command <<<
set -e
export GATK_LOCAL_JAR=~{default="/root/gatk.jar" runtime_params.gatk_override}
# Extract our data sources:
echo "Extracting data sources zip file..."
mkdir datasources_dir
tar zxvf ~{data_sources_tar_gz} -C datasources_dir --strip-components 1
DATA_SOURCES_FOLDER="$PWD/datasources_dir"
# Handle gnomAD:
if ~{use_gnomad} ; then
echo "Enabling gnomAD..."
for potential_gnomad_gz in gnomAD_exome.tar.gz gnomAD_genome.tar.gz ; do
if [[ -f ~{dollar}{DATA_SOURCES_FOLDER}/~{dollar}{potential_gnomad_gz} ]] ; then
cd ~{dollar}{DATA_SOURCES_FOLDER}
tar -zvxf ~{dollar}{potential_gnomad_gz}
cd -
else
echo "ERROR: Cannot find gnomAD folder: ~{dollar}{potential_gnomad_gz}" 1>&2
false
fi
done
fi
# Run Funcotator:
gatk --java-options "-Xmx~{runtime_params.command_mem}m" Funcotator \
--data-sources-path $DATA_SOURCES_FOLDER \
--ref-version ~{reference_version} \
--output-file-format ~{output_format} \
-R ~{ref_fasta} \
-V ~{input_vcf} \
-O ~{output_file} \
~{interval_list_arg} ~{default="" interval_list} \
--annotation-default normal_barcode:~{default="Unknown" control_id} \
--annotation-default tumor_barcode:~{default="Unknown" case_id} \
--annotation-default Center:~{default="Unknown" sequencing_center} \
--annotation-default source:~{default="Unknown" sequence_source} \
~{"--transcript-selection-mode " + transcript_selection_mode} \
~{transcript_selection_arg}~{default="" sep=" --transcript-list " transcript_selection_list} \
~{annotation_def_arg}~{default="" sep=" --annotation-default " annotation_defaults} \
~{annotation_over_arg}~{default="" sep=" --annotation-override " annotation_overrides} \
~{excluded_fields_args}~{default="" sep=" --exclude-field " funcotator_excluded_fields} \
~{filter_funcotations_args} \
~{extra_args_arg}
# Make sure we have a placeholder index for MAF files so this workflow doesn't fail:
if [[ "~{output_format}" == "MAF" ]] ; then
touch ~{output_maf_index}
fi
>>>
runtime {
docker: runtime_params.gatk_docker
bootDiskSizeGb: runtime_params.boot_disk_size
memory: runtime_params.machine_mem + " MB"
disks: "local-disk " + select_first([disk_space, runtime_params.disk]) + " HDD"
preemptible: runtime_params.preemptible
maxRetries: runtime_params.max_retries
cpu: runtime_params.cpu
}
output {
File funcotated_output_file = "~{output_file}"
File funcotated_output_file_index = "~{output_file_index}"
}
}更多教程推荐查看:GATK workflows
参考资料:
1.https://gatk.broadinstitute.org/hc/en-us
2.https://github.com/broadinstitute/gatk
