trackViewer可以用于可视化基因组数据覆盖范围和轨迹 注释 ,同时也可以用于生成棒棒糖/蒲公英图,对甲基化/突变/变异数据进行可视化,来帮助我们对多组学数据进行综合分析提供一个可靠的解决方案。
# 安装
BiocManager::install("trackViewer")1)基因信息位置绘图,下面以官方示例详细解释其用法
library(trackViewer)
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
library(org.Hs.eg.db)
# 载入演示数据
extdata <- system.file("extdata", package = "trackViewer",
mustWork = TRUE)
# importScore 用来导入BED, WIG, bedGraph 或者 BigWig文件
# importBam 可以导入Bam文件
repA <- importScore(
file.path(extdata, "cpsf160.repA_-.wig"),
file.path(extdata, "cpsf160.repA_+.wig"),
format = "WIG"
)
# 手动指定链的方向
strand(repA$dat) <- "-"
strand(repA$dat2) <- "+"
# 导入fox2基因的信息
fox2 <- importScore(
file.path(extdata, "fox2.bed"),
format = "BED",
ranges = GRanges("chr11", IRanges(122830799, 123116707))
)
dat <- coverageGR(fox2$dat)
# 手动指定fox2链的方向
fox2$dat <- dat[strand(dat) == "+"]
fox2$dat2 <- dat[strand(dat) == "-"]
# 建立染色体基因结构信息
gr <- GRanges("chr11", IRanges(122929275, 122930122), strand="-")
trs <- geneModelFromTxdb(TxDb.Hsapiens.UCSC.hg19.knownGene,
org.Hs.eg.db,
gr=gr)
# 获取FMR1基因信息
entrezIDforFMR1 <- get("FMR1", org.Hs.egSYMBOL2EG)
theTrack <- geneTrack(entrezIDforFMR1,TxDb.Hsapiens.UCSC.hg19.knownGene)[[1]]
# 设置track样式
viewerStyle <- trackViewerStyle()
setTrackViewerStyleParam(viewerStyle, "margin", c(.1, .05, .02, .02))
vp <- viewTracks(
# 这里可以设置很多具体参数,可以查看文档修改
trackList(repA, fox2, trs),
gr = gr,
viewerStyle = viewerStyle,
autoOptimizeStyle = TRUE
)
# 添加标注
addGuideLine(c(122929767, 122929969), vp = vp)
addArrowMark(list(x = 122929650,
y = 2),
# 2 means track 2 from the bottom.
label = "label",
col = "blue",
vp = vp)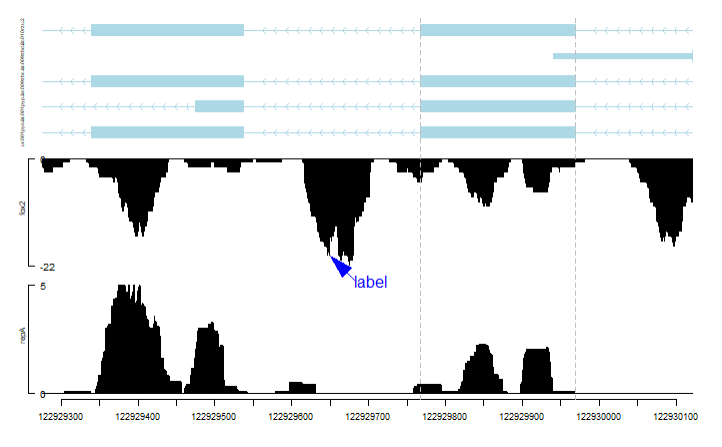
我们也可以简单设置下颜色( 续上面代码 ):
# theme 参数选择主题
optSty <- optimizeStyle(trackList(repA, fox2, trs), theme = "col")
trackList <- optSty$tracks
viewerStyle <- optSty$style
vp <- viewTracks(trackList, gr = gr, viewerStyle = viewerStyle)
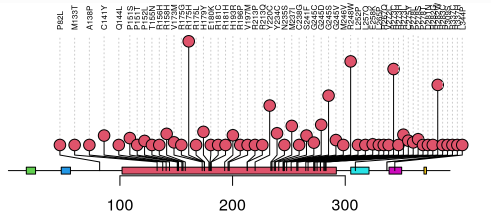
2)SNP棒棒糖图绘制:
library(trackViewer)
# SNP位置信息
SNP <- c(10, 100, 105, 108, 400, 410, 420, 600, 700, 805, 840, 1400, 1402)
# 染色体信息
sample.gr <-
GRanges("chr1", IRanges(SNP, width = 1, names = paste0("snp", SNP)))
features <- GRanges("chr1", IRanges(
c(1, 501, 1001),
width = c(120, 400, 405),
names = paste0("block", 1:3)
))
# 设置填充色
features$fill <- c("#FF8833", "#51C6E6", "#DFA32D")
# 设置棒棒糖点的颜色
sample.gr$color <- sample.int(6, length(SNP), replace=TRUE)
sample.gr$border <- sample(c("gray80", "gray30"), length(SNP), replace=TRUE)
sample.gr$alpha <- sample(100:255, length(SNP), replace = TRUE)/255
# 添加标签
sample.gr$label <- as.character(1:length(sample.gr))
sample.gr$label.col <- ifelse(sample.gr$alpha>0.5, "white", "black")
# 更改区块高度
features$height <- c(0.02, 0.05, 0.08)
# 绘图
lolliplot(sample.gr, features, jitter="label")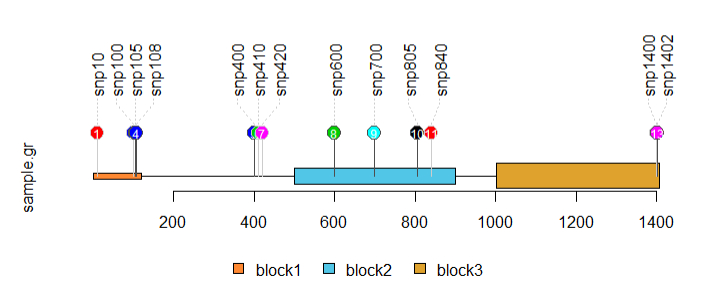
3)绘制蒲公英图
library(trackViewer)
# 演示数据
SNP <-
c(10, 100, 105, 108, 400, 410, 420, 600, 700, 805, 840, 1400, 1402)
SNP.gr <-
GRanges("chr1",
IRanges(SNP, width = 1, names = paste0("snp", SNP)),
score = sample.int(100, length(SNP)) / 100)
features <- GRanges(
"chr1",
IRanges(
c(1, 501, 1001),
width = c(120, 500, 405),
names = paste0("block", 1:3)
),
color = "black",
fill = c("#FF8833", "#51C6E6", "#DFA32D"),
height = c(0.1, 0.05, 0.08)
)
# 设置属性
SNP.gr$color <- 3
SNP.gr$border <- "gray"
m <- max(SNP.gr$score)
SNP.gr$score <- SNP.gr$score/m
# 绘图
dandelion.plot(SNP.gr, features, type = "fan")

trackViewer的功能还是很强大的,还支持诸多基因组图形展示,具体内容可以查看参考资料。
参考资料:
1.http://bioconductor.org/packages/release/bioc/vignettes/trackViewer/inst/doc/trackViewer.html

载入演示数据 extdata
extdata,这个文件的格式文件可以给下吗?谢谢