DEP为蛋白质组数据分析提供了一个可靠的分析流程,它需要由原始质谱数据的定量分析软件(如MaxQuant或IsobarQuant)生成的结果文件作为输入(例如txt文件),包含了数据预处理、数据过滤、缺失值计算、差异分析等功能。
同时提供可视化工具来探索结果,包括heatmap, volcano plot和barplot等。对于在R方面不了解的研究人员,DEP还包含完整的分析工作流和生成报告(基于shiny的可视化操作),非常方便使用。
1)安装( 需要安装Pandoc,以及用于Windows的Rtools和用于Linux的NetCDF )
# 通过BiocManager安装
if (!requireNamespace("BiocManager", quietly=TRUE))
install.packages("BiocManager")
BiocManager::install("DEP")
library("DEP")2)DEP支持交互式分析(目前支持label free分析和TMT数据分析),按照如下方式启动:
# For LFQ analysis
run_app("LFQ")
# For TMT analysis
run_app("TMT")
3)具体分析流程,按照如下方式
3.1 加载测试数据
library("dplyr")
# 测试数据
data <- UbiLength
# 数据过滤,过滤污染物蛋白质和decoy数据
data <- filter(data, Reverse != "+", Potential.contaminant != "+")
3.2 数据预处理
# 重复基于名称处理
data$Gene.names %>% duplicated() %>% any()
# 查看重复的基因
data %>% group_by(Gene.names) %>% summarize(frequency = n()) %>%
arrange(desc(frequency)) %>% filter(frequency > 1)
# 确保基因名称唯一
data_unique <- make_unique(data, "Gene.names", "Protein.IDs", delim = ";")
# 验证
data$name %>% duplicated() %>% any()3.3 生成一个 SummarizedExperiment 对象,方便后续处理
# 生成SummarizedExperiment对象
LFQ_columns <- grep("LFQ.", colnames(data_unique)) # get LFQ column numbers
experimental_design <- UbiLength_ExpDesign
data_se <- make_se(data_unique, LFQ_columns, experimental_design)
LFQ_columns <- grep("LFQ.", colnames(data_unique)) # get LFQ column numbers
data_se_parsed <- make_se_parse(data_unique, LFQ_columns)
# 查看
data_se
## class: SummarizedExperiment
## dim: 2941 12
## metadata(0):
## assays(1): ''
## rownames(2941): RBM47 UBA6 ... ATXN2.3 X6RHB9
## rowData names(13): Protein.IDs Majority.protein.IDs ... name ID
## colnames(12): Ubi4_1 Ubi4_2 ... Ubi1_2 Ubi1_3
## colData names(4): label ID condition replicate3.4 缺失值过滤
# 查看蛋白鉴定数
plot_frequency(data_se)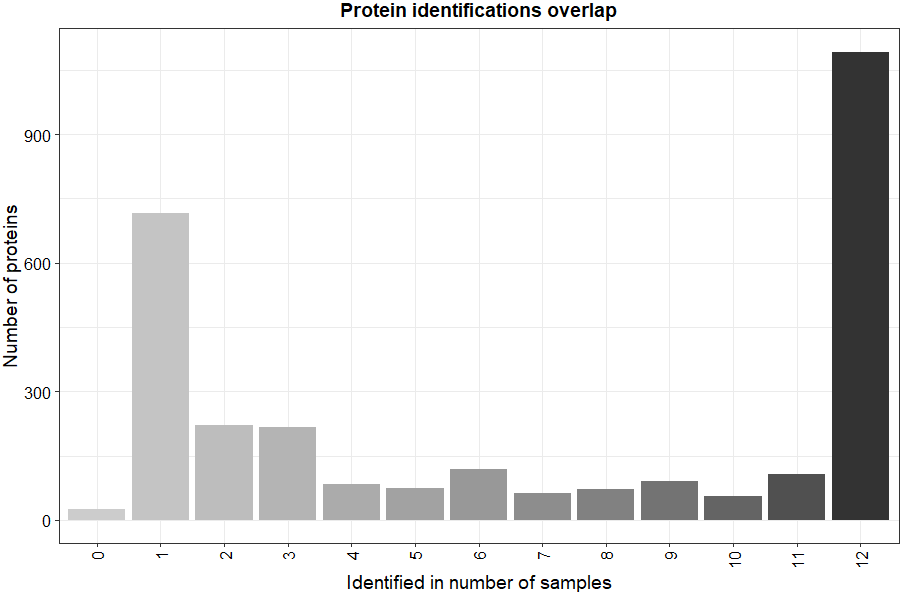
# 过滤缺失值,至少在一组中全部被鉴定到
data_filt <- filter_missval(data_se, thr = 0)
# 过滤缺失值,至少在一组中少一个(如一组3个,其中2个鉴定到该蛋白)被鉴定到
data_filt2 <- filter_missval(data_se, thr = 1)plot_numbers(data_filt) # 查看蛋白数量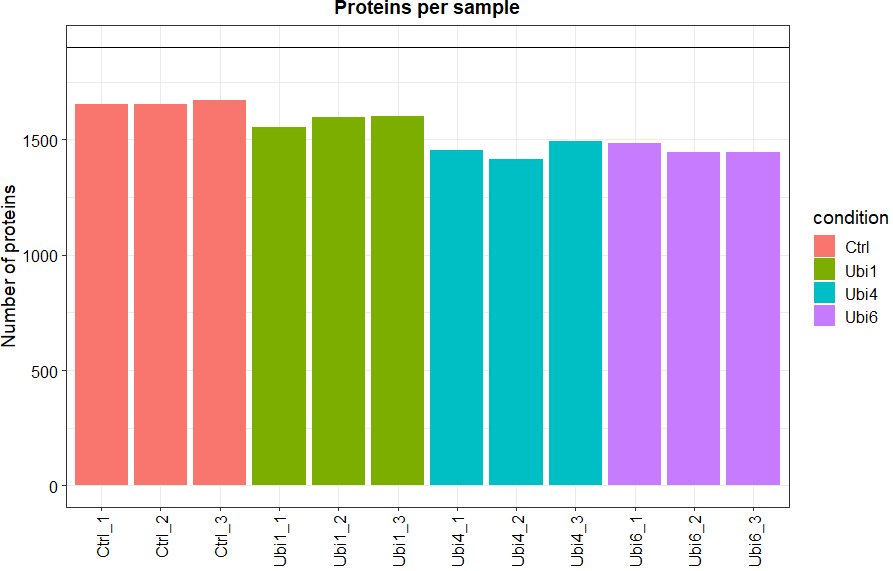
plot_coverage(data_filt) # 绘制样品间蛋白质鉴定重叠的柱状图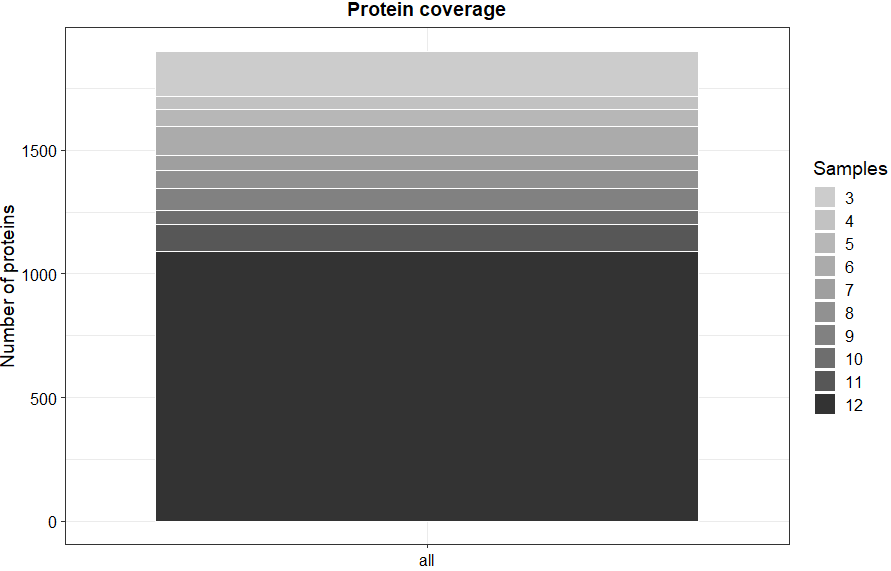
3.5 数据标准化
data_norm <- normalize_vsn(data_filt)
# 绘图
plot_normalization(data_filt, data_norm)
3.6 缺失值填充
# 查看缺失值情况
plot_missval(data_filt)
plot_detect(data_filt) # 缺失值检测,有或没有缺失值的蛋白质的概率密度曲线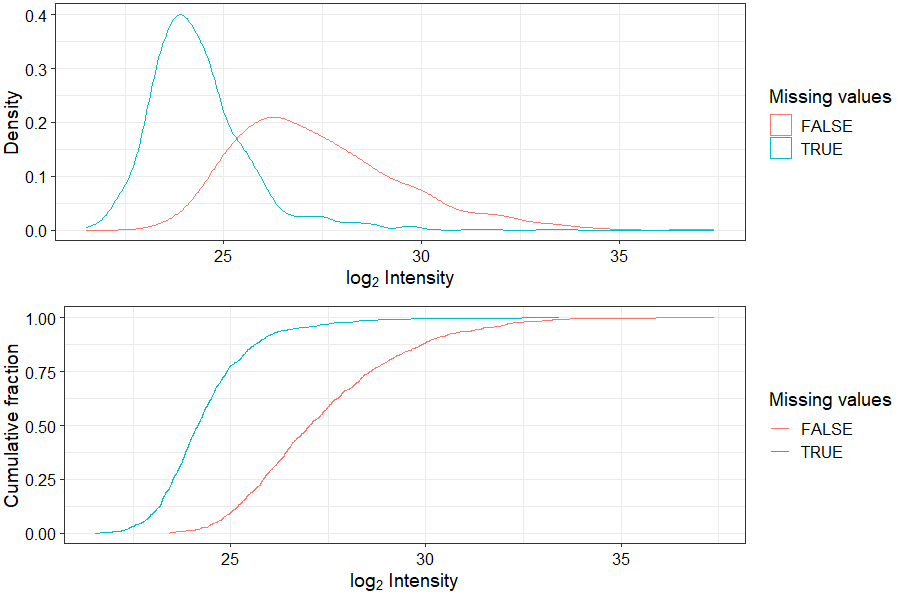
# MNAR(非随机缺失)缺失值填充
data_imp <- impute(data_norm, fun = "MinProb", q = 0.01)
data_imp_man <- impute(data_norm, fun = "man", shift = 1.8, scale = 0.3)
data_imp_knn <- impute(data_norm, fun = "knn", rowmax = 0.9)# 绘图查看填充效果
plot_imputation(data_norm, data_imp)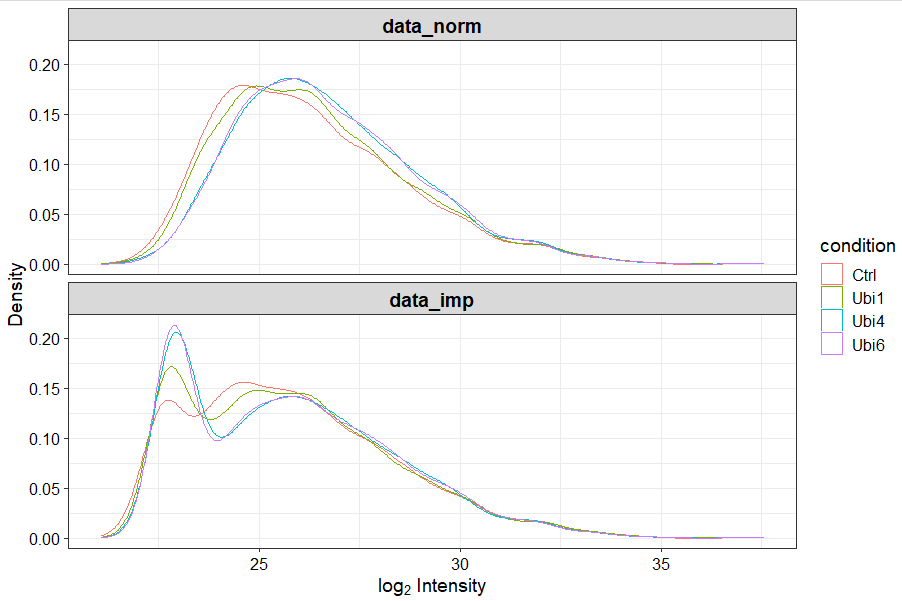
# 绘图查看knn填充效果
plot_imputation(data_norm, data_imp_knn)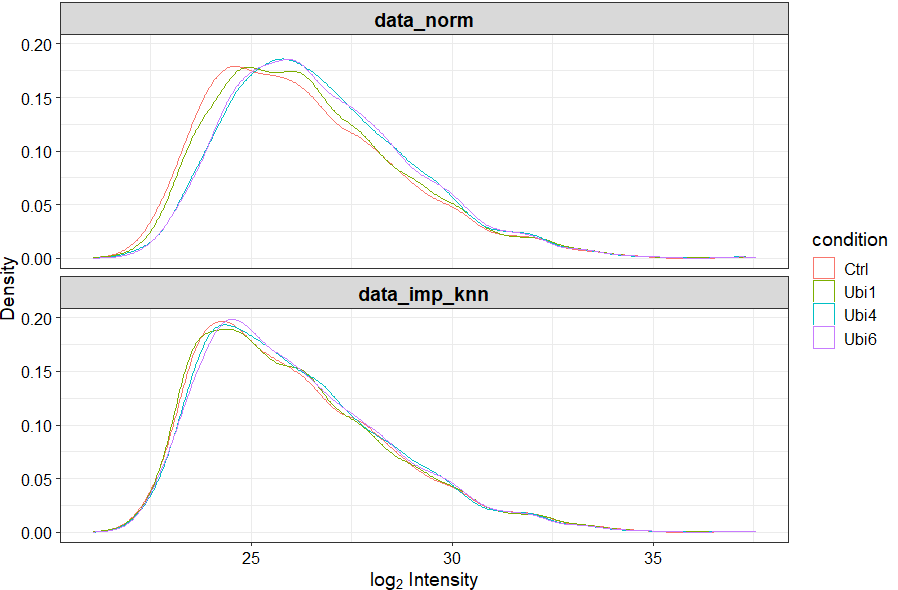
3.7 差异分析
# 每一个样本vs对照
data_diff <- test_diff(data_imp, type = "control", control = "Ctrl")
# 测试所有可能的比较
data_diff_all_contrasts <- test_diff(data_imp, type = "all")
# 手动指定比较分组
data_diff_manual <- test_diff(data_imp, type = "manual",
test = c("Ubi4_vs_Ctrl", "Ubi6_vs_Ctrl"))
# 添加阈值,筛选
dep <- add_rejections(data_diff, alpha = 0.05, lfc = log2(1.5))3.8 结果可视化
# 主成分分析
plot_pca(dep, x = 1, y = 2, n = 500, point_size = 4)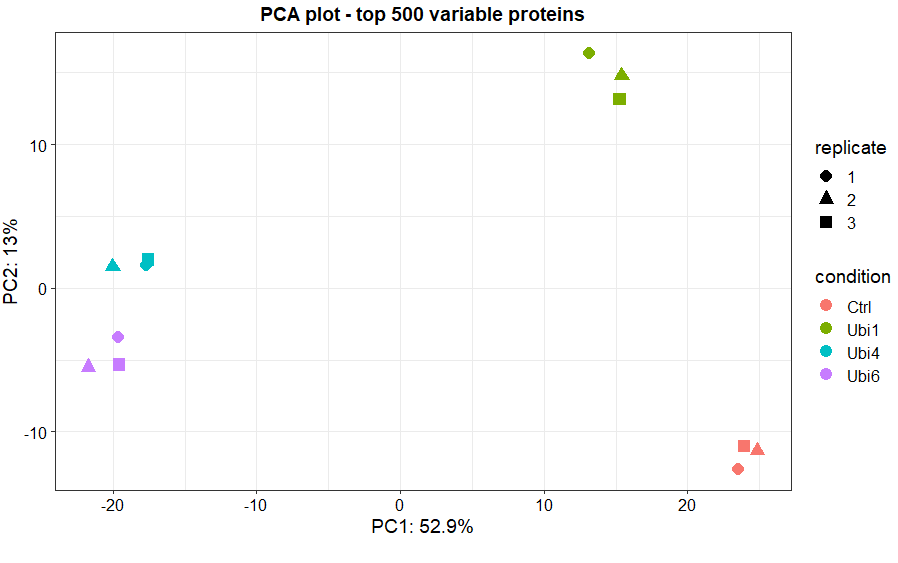
# 相关性分析
plot_cor(dep, significant = TRUE, lower = 0, upper = 1, pal = "Reds")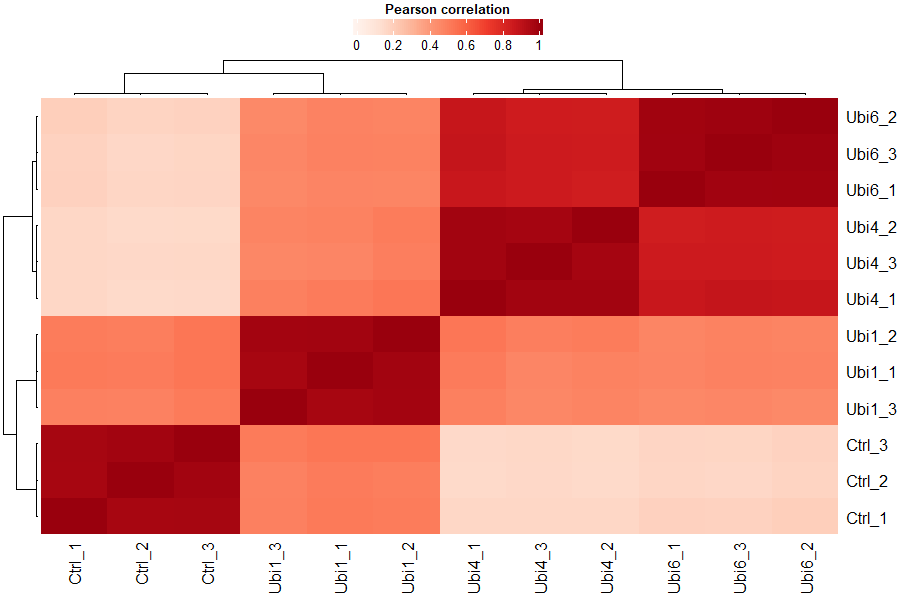
# 热图
plot_heatmap(dep, type = "centered", kmeans = TRUE,
k = 6, col_limit = 4, show_row_names = FALSE,
indicate = c("condition", "replicate"))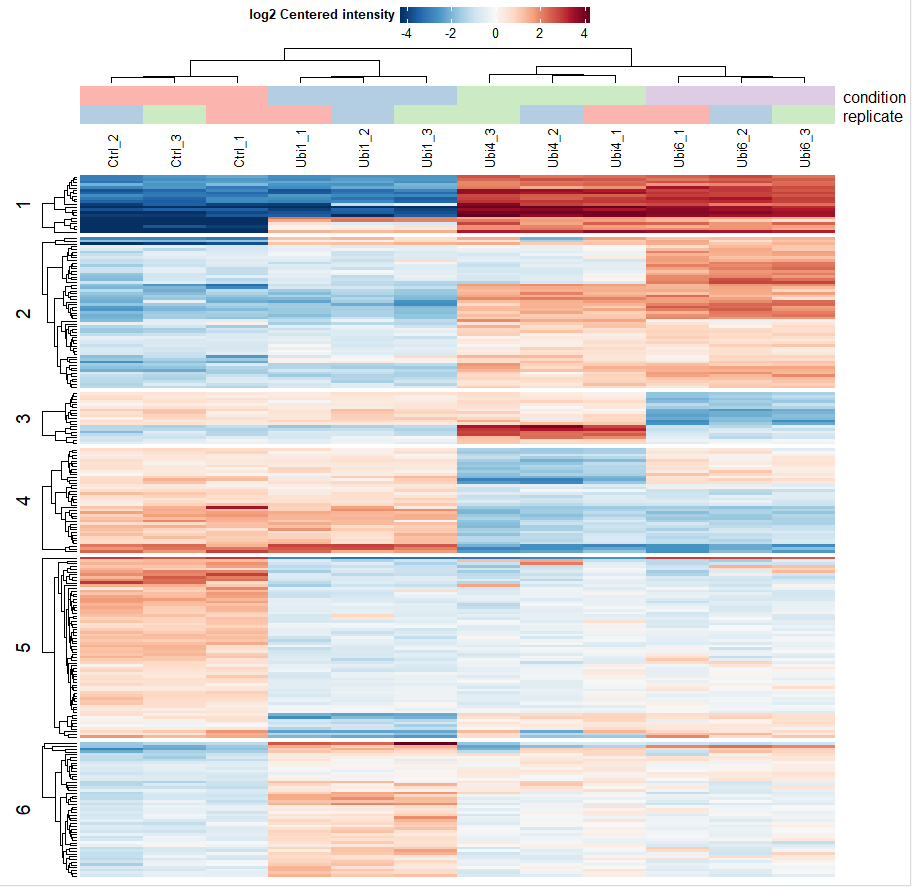
# 火山图
plot_volcano(dep, contrast = "Ubi6_vs_Ctrl", label_size = 2, add_names = TRUE)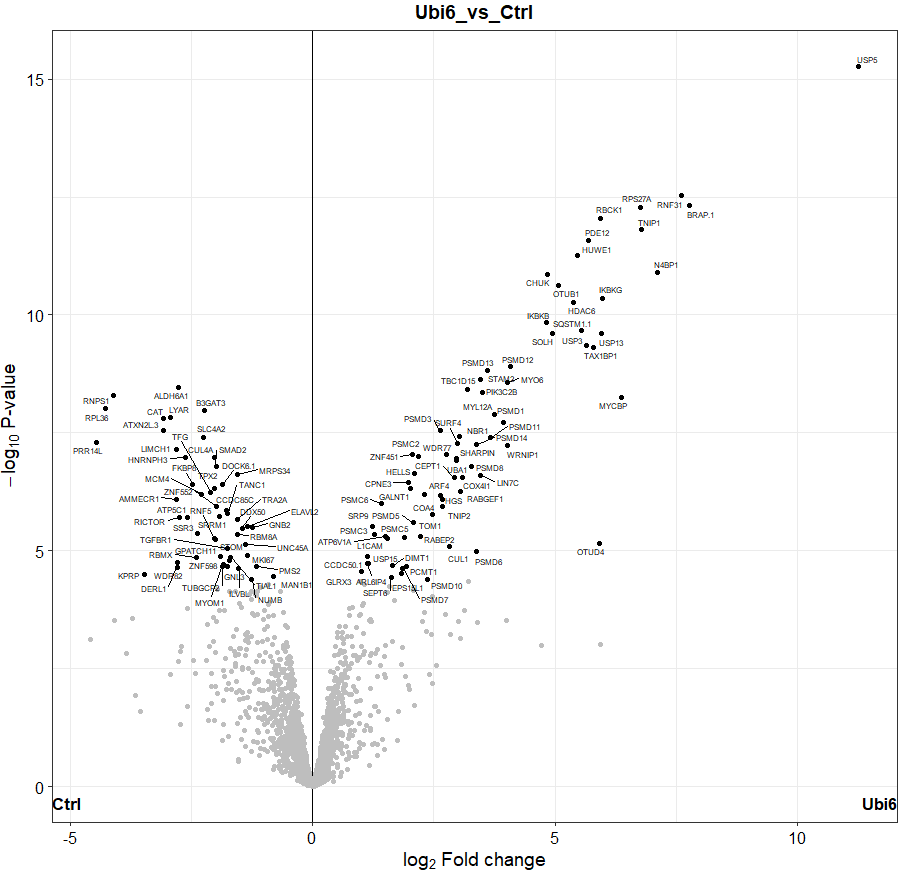
3.9 个性化显示部分感兴趣的基因/蛋白
# 条形图
plot_single(dep, proteins = c("USP15", "IKBKG"))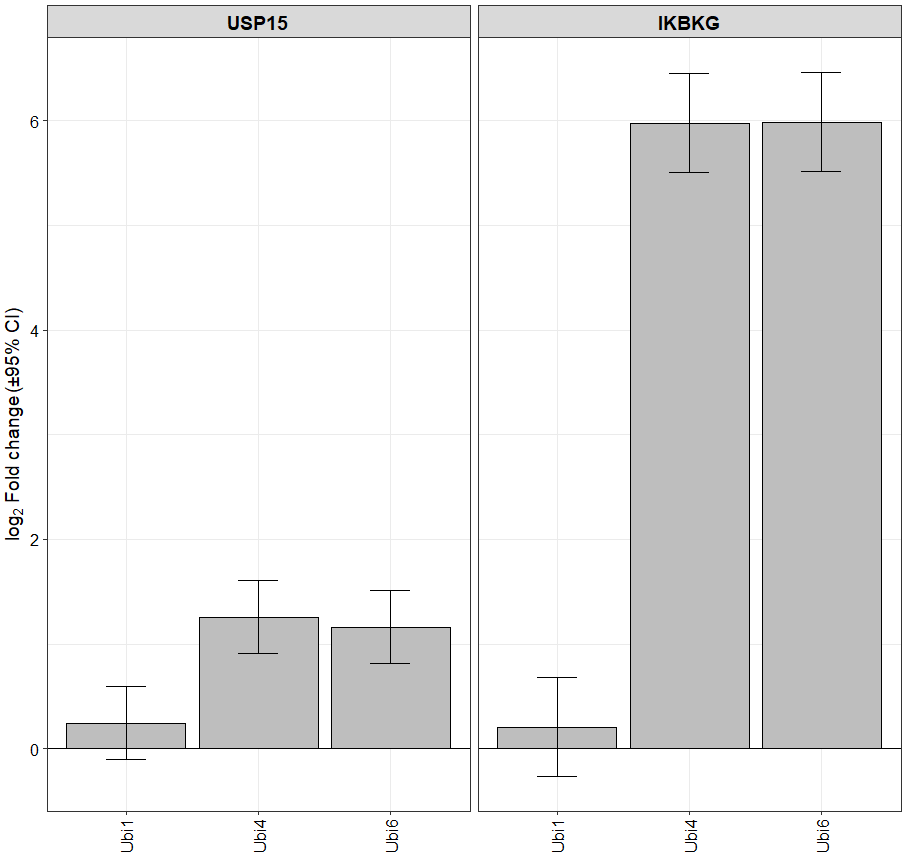
plot_single(dep, proteins = "USP15", type = "centered")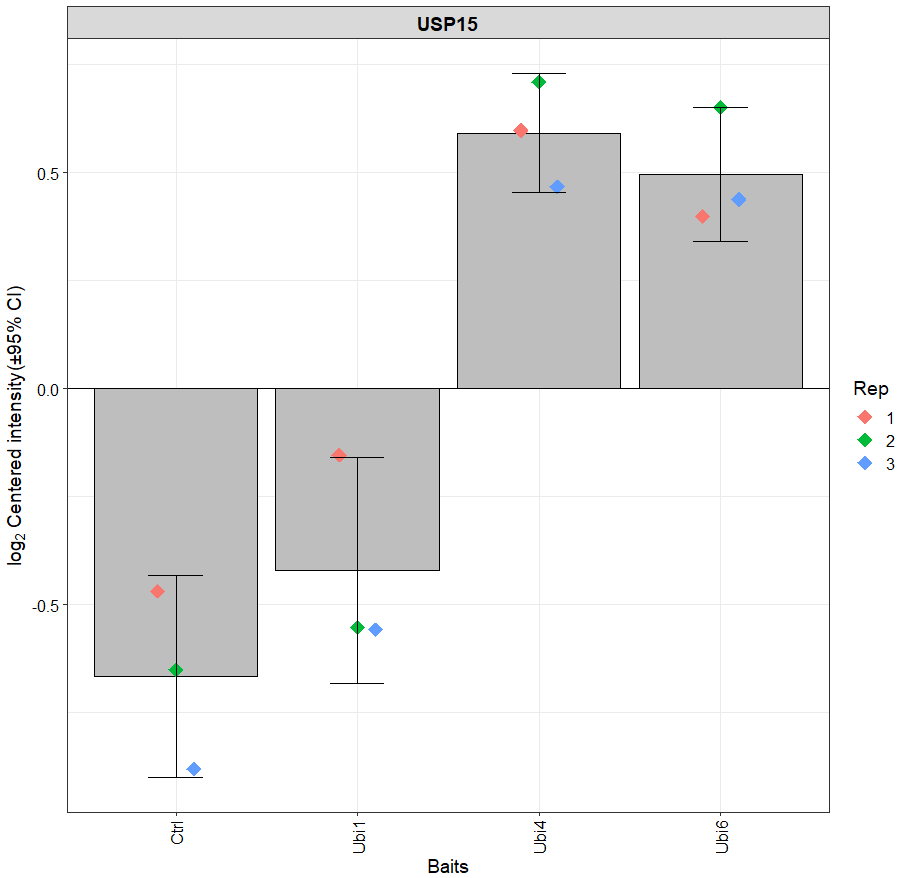
# 绘制不同条件下显著蛋白的频率图
plot_cond(dep)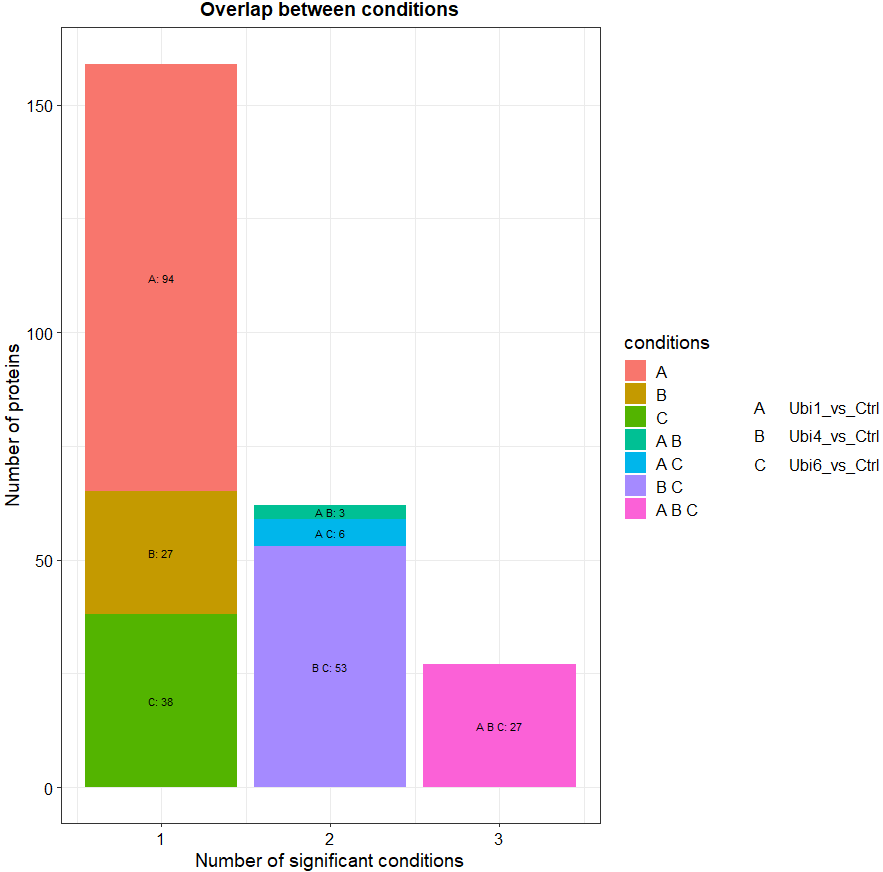
至此,蛋白组学的基本分析结束了,后续的分析包括GO/KEGG、IPA等分析下一篇在详细讲解。
参考资料:
1.http://www.bioconductor.org/packages/release/bioc/vignettes/DEP/inst/doc/DEP.html#lfq-based-dep-analysis

DDL
可以在重要蛋白热图中添加蛋白名称吗?代码是?谢谢
陈浩
可以用ggrepel包辅助显示蛋白名称,示例如下(label.data是你想标注的蛋白信息数据框):
plot + geom_label_repel(data = label.data,
aes(x = logFC, y = -log10(adj.P.Val), label = label),
size = 4,
color=”black”,
box.padding = unit(0.4, “lines”),
segment.color = “black”,
segment.size = 0.4,
)
myy
您好,请问experimental_design <- UbiLength_ExpDesign这里我自己的数据的话,应该怎么得到experimental_design ,打扰了,谢谢
正规表现
您好,在使用run-LFQ报错:正规表现’^[‘不对,原因是’Missing ‘]”,请问是什么原因呢?
陈浩
请问你使用的是那个版本的DEP呢?
求指导
3.3步骤就一直走路下去了
陈浩
具体是遇到什么问题呢?
留白
请问如果没有生物学重复的样本可以分析吗
陈浩
没有生物学重复的不建议用它来分析。一般来说没有生物学重复的样本建议可以通过差异倍数来进行初筛(这个一般用于摸索实验条件),然后设计实验用更多样本来做验证,对于非稀有样本还是推荐添加生物重复来证实结论的扩展性。
孙小文
3.5的绘图部分,3.6查看缺失值情况,代码有误。
陈浩
谢谢指出,具体是什么错误了?你安装的包的版本号是多少?